Die Desorption insbesondere der ersten Adsorbatschichten der Metalle Kupfer und Silber von einer Re(0001)-Oberfläche stellt einen Vorgang dar, dessen Mechanismus nicht trivial ist. Aus der Form der TD-Spektren, den daraus gewonnenen Orderplots, und dem Verlauf der Desorptionsenergie können Hinweise auf die Ausbildung von Oberflächen-Phasen und Phasenübergängen während der Desorption entnommen werden. Beim Übergang zwischen Lagen kommt es zu starken Wechselwirkungen, die im Gittermisfit der Systeme (Cu: -7,6%, Ag: +4,6%) begründet sind. Es werden die kritischen Parameter sowie die interne Wechselwirkungsenergie der Adteilchen bestimmt, und es wird das Desorptionsverhalten anhand des Modells des zweidimensionalem Gases beschrieben.
1. Einleitung
2.1 Das Modell des zweidimensionalen Gases
2.3 Die Bragg-Williams-Näherung (BWA)
2.4 Desorptionsratengleichungen
4.1 Flanken-Methode
Phasenübergänge in Metall-Adsorbatsystemen sind seit etwa zwanzig Jahren gut bekannt und untersucht worden. Hervorzuheben sind hier die Arbeiten von Bauer et al. [B74/1, B75/1]. Im besonderen Interesse standen die Systeme Ni/W(110), Cu/W(110), Ag/W(110) [B86/2], Cu/Mo(110) [B87/1], Au/Mo(110) [B88/1]. Durch Thermodesorptionsspektroskopie, Isothermaldesorptionsspektroskopie und Messungen der Elektronenaustrittsarbeitsänderung wurden solche Phasengrenzen untersucht. Des weiteren wurden viele theoretische Arbeiten zu diesem Thema veröffentlicht, z.B. von Bauer et al. [B85/1], Kreuzer/Payne [K89/1], Nagai [N86/1], Zhdanov [Z81/1]. Als thermodynamische Näherungen wurden dabei die Bragg-Williams-Näherung, die Quasichemische Näherung, das Isingmodell u.a. verwandt.
Die Re(0001)-Oberfläche eignet sich besonders gut für die Thermodesorptionsuntersuchungen, da das Rhenium einerseits einen sehr hohen Schmelzpunkt von 3453K andererseits eine besonders glatte Oberfläche besitzt: die atomare Korrugation beträgt etwa 0,1Å [PAR96]. Kupfer und Silber als Adsorbatmaterialien bieten sich wegen ihrer niedrigen Schmelzpunkte von 1358K (Cu) bzw. 1234K (Ag) an. Legierungsbildung zwischen den Adsorbatmetallen und dem Rhenium ist nicht zu erwarten. Kupfer und Silber kristllisieren im fcc-Gitter. Nur sehr genaue und umfangreiche Messungen, Auswertungen und Interpretationen von TD-Spektren können detaillierte Informationen über das Verhalten der Systeme bei Desorptionstemperaturen geben. Ein Anzeichen für einen Phasenübergang ist beispielsweise der Wechsel der Desorptionsordnung, wie er bei unseren Systemen beobachtet werden konnte. Aber auch aus dem Verlauf der Desorptionsenergie können Rückschlüsse auf die Wechselwirkungen der Adteilchen gezogen werden. Augerelektronenspektren liefern ebenfalls Hinweise zu Phasenübergängen unserer Systeme.
Im folgenden wird zunächst das Modell des zweidimensionalen Gases vorgestellt, mit dem der Desorptionsmechanismus erklärt wird. Unter Verwendung der Bragg-Williams-Näherung wird das Desorptionsverhalten der Systeme beschrieben. Im darauffolgenden experimentellen Teil wird kurz auf die Durchführung der Experimente eingegangen. Weiterhin wird gezeigt, wie sich die zur Charakterisierung der Phasenübergänge nötigen Informationen in den Meßergebnissen dokumentieren. Daraufhin wird die Messung der Phasengrenze beschrieben. Abschließend werden die Ergebnisse zusammengefaßt und diskutiert.
![]()
2.1Das Modell des zweidimensionalen Gases
Adsorptionssysteme mit einer glatten Oberfläche (wie die Re(0001)-Oberfläche) können besonders gut mit Hilfe des Modells des 2D-Gases beschrieben werden. Dieses Modell basiert auf dem Gleichgewicht zwischen zwei Anteilen, die sich auf der Oberfläche befinden. Der eine ist in Adsorbatinseln lokalisiert, der andere kann sich auf der blanken Substratoberfläche frei bewegen und wird als 2D-Gas angesehen, das ideale Eigenschaften besitzen soll.
Die Re(0001)-Oberfläche mit ihrer genau definierten Anzahl von
Adsorptionsplätzen N ist, je nach Verlauf der Adsorption, mit einer
bestimmten Anzahl (![]() ) von Adsorbatteilchen
bedeckt. Diese gehen sowohl Wechselwirkungen mit dem Substrat (gekennzeichnet
durch das Potential in z-Richtung Vz) als auch mit ihren nächsten
Nachbarn ein, abhängig von ihrer Oberflächenkonzentration und
Koordination, (bestimmt durch die interne Wechselwirkungsenergie EWW).
Unser Modell beschränkt sich auf die Vorgänge innerhalb einer
Lage. Damit kann man die Teilchendichte durch den Bedeckungsgrad Q
=NA/N ausdrücken. Q kann maximal
1 sein, wenn jeder Adsorptionsplatz durch genau ein Teilchen besetzt ist.
) von Adsorbatteilchen
bedeckt. Diese gehen sowohl Wechselwirkungen mit dem Substrat (gekennzeichnet
durch das Potential in z-Richtung Vz) als auch mit ihren nächsten
Nachbarn ein, abhängig von ihrer Oberflächenkonzentration und
Koordination, (bestimmt durch die interne Wechselwirkungsenergie EWW).
Unser Modell beschränkt sich auf die Vorgänge innerhalb einer
Lage. Damit kann man die Teilchendichte durch den Bedeckungsgrad Q
=NA/N ausdrücken. Q kann maximal
1 sein, wenn jeder Adsorptionsplatz durch genau ein Teilchen besetzt ist.
Bei geringen Bedeckungen oder hohen Temperaturen sind alle Teilchen auf der Oberfläche delokalisiert und bilden eine reine 2D-Gasphase aus. Bei einem bestimmten (Grenz-) Bedeckungsgrad Q g der von der Temperatur abhängt, werden die attraktiven Wechselwirkungen wegen der zunehmenden räumlichen Nähe der Teilchen sehr stark. Damit nimmt auch die Wahrscheinlichkeit zur Bildung von stabilen Kondensationskeimen zu. Dies hat zur Folge, daß eine Mischphase gebildet wird, die aus kondensierten und 2D-Gasteilchen besteht. Die kondensierten Teilchen bilden Inseln auf der Oberfläche aus, zwischen denen sich weiterhin 2D-Gasteilchen bewegen können. Unterhalb der kritischen Temperatur Tc stehen in der Mischphase die Kondensat- und 2D-Gasteilchen im Gleichgewicht. Bei gleichbleibender Temperatur entspricht die 2D-Gaskonzentration derjenigen bei Q g. Die gebildeten Inseln sind in sich kompakt, was nichts über ihre äußere Form aussagt. Die Aktivität der Teilchen in den Inseln ist somit (wie im dreidimensionalen Fall) eins. Die Bedeckung mit kondensierten Teilchen Q c kann sich jedoch durch die Veränderung der Anzahl und der Größe der Inseln ändern. Durch einen steigenden Anteil von Kondensatteilchen (etwa bei Adsorption) wird der dem 2D-Gas zur Verfügung stehende Raum kleiner, weshalb sich auch bei konstanter Gasdichte der Flächenanteil dieser Spezies verringert.
Wendet man die Thermodesorptionsspektroskopie zur Untersuchung von Adsorbatsystemen an, werden diese in dem erhöhten Temperaturbereich vom Einsetzen bis zum Abklingen der Desorption betrachtet. Die Desorption wird durch thermische Energie kBT, die dem System über eine lineare Heizrampe zugeführt wird, induziert. Zu Beginn der Desorption ist die Bedeckung gleich der Anfangsbedeckung Q 0 und nimmt während des Prozesses ab, bis schließlich kein Teilchen mehr auf der Substratoberfläche vorhanden ist. (Die Restbedeckung zu einem bestimmten Zeitpunkt soll deshalb mit Q bezeichnet werden.) Die Energie, die aufgebracht werden muß, damit ein (Mol) Teilchen desorbiert, ist die Desorptionsenergie D Edes.
Die Desorption soll nach unserem Modell nur aus der 2D-Gasphase mit der Rate r3D möglich sein. Im Bereich sehr kleiner Bedeckungen sind nur 2D-Gasteilchen vorhanden, die direkt desorbieren können. Sind sowohl Gas- als auch Kondensatteilchen vorhanden, gehen diese zunächst mit rcg (cond. - 2D-Gas) in die 2D-Gasphase über und von dort in die 3D-Gasphase [K91/3, K88/4].
Dieser Übergang ins 2D-Gas ist so schnell (rcg>>r3D), daß ein Quasigleichgewicht zwischen den beiden Phasen besteht. Solange kondensierte Teilchen vorhanden sind, werden durch das o.a. vorgelagerte Gleichgewicht 2D-Gasteilchen sofort nachgebildet, wodurch ihre Konzentration konstant bleibt. (Diffusionsprozesse sind wegen ihrer hohen Geschwindigkeit nicht ratenbestimmend.) Wir können die Vorgänge bei der Erwärmung eines solchen Adsorbatsystems also über ein Schema approximieren, das die Desorption als eine Reaktion mit vorgelagertem Gleichgewicht beschreibt.
2.3 Die Bragg-Williams-Näherung(BWA)
Als ein wegen seiner mathematischen Einfachheit gut zu überschauendes Modell hat sich die Bragg-Williams-Näherung (BWA) bewährt [SPA85]. Mit ihrer Hilfe lassen sich bestimmte Zusammenhänge besser verstehen. Zunächst wird am allgemeinen Fall der Inhalt der Näherung beschrieben. Danach wird der Bezug zu unseren Adsorbatsystemen hergestellt. Es gelten folgende Voraussetzungen:
Diese Näherungen sind beim hier gewählten System recht gut erfüllt, wenn der Bedeckungsgradbereich nicht ganz bis zur Auffüllung der Monolage betrachtet wird. Dort können Wirkungen der Gitterfehlanpassungen (-7,6% (Cu) bzw. +4,6% (Ag)) und einer eventuellen Besetzung der zweiten Lage sichtbar werden.
Es existieren also 1/2cQ N Adteilchenpaare auf der Oberfläche. Die Wechselwirkung eines Teilchens A mit dem restlichen "Molekularfeld" ist 1/2cQ EAA, die einer Teilchenart folglich 1/2cNQ EAA. Die laterale Wechselwirkungsenergie des betrachteten Adsorbatteilchens mit seinen nächsten Nachbarn kann attraktiv (EAA<0) oder repulsiv (EAA>0) sein. Treten zwei Teilchenarten auf, z.B. A und B, ergibt sich
NA=Q N, NB=(1-Q
)N und ![]() (1)
(1)
und für die dabei nötige Energiekorrektur
![]() (2)
(2)
für ein Teilchen.
Die Zustandssumme für ein solches System besteht aus einem präexponentiellen Teil, der die Verteilung der Teilchen berücksichtigt, und einem Exponentialteil, der die energetischen Wechselwirkungen der Paare AA, BB, AB beschreibt [SPA85].
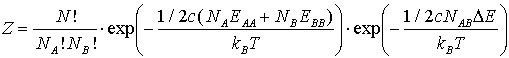 (3)
(3)
Nach einigen Umformungen und unter Betrachtung eines Entmischungsvorganges erhält man das chemische Potential für das Gesamtsystem
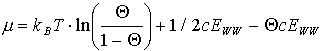 (4)
(4)
Bei der Anwendung der BWA auf unsere Adsorbatsysteme , bei denen jeweils nur eine Adteilchenart vorhanden ist, wird NA mit der Anzahl der Adteilchen identifiziert und NB mit der Zahl leerer Gitterplätze. Eine ideale Mischung von Adteilchen NA und leeren Plätzen NB entspricht damit der idealen Verteilung in Form eines 2D-Gases. Eine Entmischung steht für die Ausbildung von Adsorbatinseln (kondensierte Teilchen), d.h. für das Auftreten einer zweiten Phase. EWW ist also zunächst eine Stabilisierungsenergie für die Ausbildung der Mischphase.
Allerdings kann man hier EWW zusätzlich als eine Wechselwirkungsenergie auffassen. Dazu wird in Betracht gezogen, daß sowohl die Wechselwirkung zweier leerer Gitterplätze miteinander Null ist (EBB=0), als auch die solcher Gitterplätze mit Adteilchen (EAB=0). Es kommt immer dann zur Ausbildung des Mischphasengebietes, wenn EWW positiv ist, was einer attraktiven Wechselwirkung entpricht (-EWW=EAA<0). Gemäß m =m h+m s läßt sich auch das chemische Potential in einen energetischen und entropischen Anteil zerlegen:
![]()
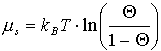 (5)
(5)
Auf Grund des unterschiedlichen Verlaufs des energetischen und des entropischen Anteils mit dem Bedeckungsgrad besitzt die Funktion des chemischen Potentials als Summe der beiden Anteile drei Nullstellen. Für m=0 bildet sich ein Gleichgewicht zwischen Bildung und Auflösung von Inseln (dem Entmischungs- und Mischungsvorgang), also das Phasengleichgewicht aus. Das bedeutet, daß der energetische Beitrag zur Ablösung der Adteilchen von Inseln vom entropischen Beitrag kompensiert wird. Dies ist erfüllt, wenn -m s=m h ist. Für die Phasengrenze gilt also:
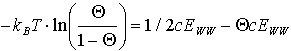 (6)
(6)
Es ergibt sich bei Q =0.5 ein spiegelsymmetrischer Verlauf der Phasengrenze. (Bei der Berücksichtigung von z.B. einer Dreikörper-Wechselwirkung geht dieser symmetrische Verlauf verloren [Z86/4].) Die Breite der Koexistenzphase zu einer bestimmten Temperatur ist dabei durch die Lage der Nullstellen von m bei eben dieser Temperatur gegeben.
Aus der Phasengrenzbedingung läßt sich EWW bestimmen, indem man Q®1/2 und T=TC setzt (TC=kritische Temperatur der Phase) [B87/1, N84/1, N85/1]. Es ergibt sich der allgemeine Zusammenhang:
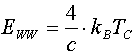 (7)
(7)
2.4 Desorptionsratengleichungen
Das chemische Gesamtpotential läßt sich als Differenz des chemischen Potentials des Endzustandes des Mischvorganges (das des Misch- bzw. Zweiphasengebietes (2)) und dessen Anfangszustandes (das der reinen Phase (1)) (m =-m 1+m 2) darstellen [K88/1, K89/1, K91/3, N84/1, N88/1]. Nur im Einphasengebiet kann Q einer konkreten Spezies zugeordnet werden (Q 1=Q g (bzw. Q 1=Q s), aber Q 2=Q c+Q g)
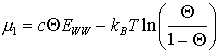
![]() (8)
(8)
Eine Desorptionsratengleichung
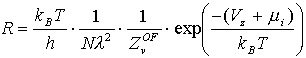 , (9)
, (9)
in der die gesamte Q -Abhängigkeit in m enthalten ist [B87/1, K91/3], kann in Anteile für die beiden Bereiche (1) und (2) aufgeteilt werden [N86/1, N88/1]. Hier ist die makroskopische Meßgröße "Desorptionsenergie" durch die mikroskopischen Größen "Oberflächenpotential in z-Richtung" und "chemisches Wechselwirkungspotential der Adteilchen" ersetzt worden D Edes=Vz+m i (l =thermische Wellenlänge).
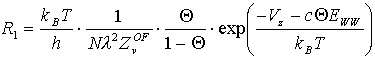 (10)
(10)
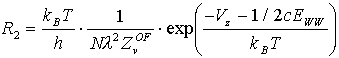 (11)
(11)
Mit diesen Gleichungen lassen sich sowohl der Verlauf der Desorptionsenergie als auch der der isothermen Desorptionsrate als Funktionen des Bedeckungsgrades beschreiben.
Auf den genauen Aufbau der Meßapparatur sowie die Meßmethoden soll hier nicht weiter eingegangen werden. Wir verweisen in diesem Zusammenhang auf frühere Arbeiten, insbesondere [S98/1] und [W98/1].
Sowohl bei der Verwendung von Kupfer als auch Silber als Adsorbat wurde derselbe Rhenium-Einkristall in dergleichen Apparatur verwandt. Die Deposition der Adsorbatmetalle geschah durch atomare Verdampfung der Metalle aus einer Knudsenzelle. Die bei der Desorption verwendete lineare Heizrampe wurde über einen Computer gesteuert, mit dessen Hilfe auch die Meßdaten erfaßt worden sind. Die Kristalltemperaturen betrugen J =390°C (Kupfer), bzw. J =470°C (Silber) während des Aufgedampfens, und es wurde mit einer Heizrate von b =7,7K/s (Kupfer), bzw. b =2,5K/s (Silber) desorbiert.
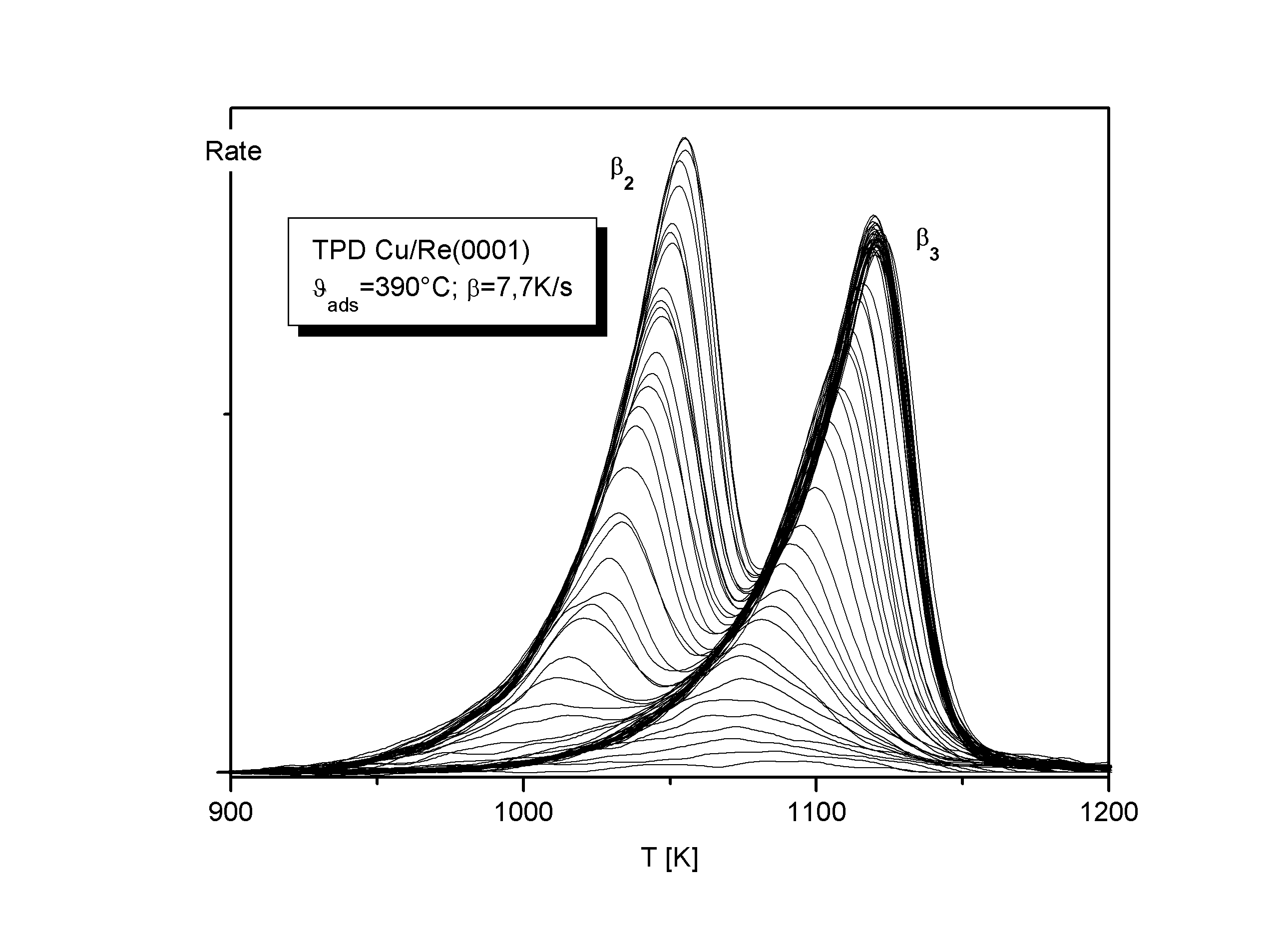
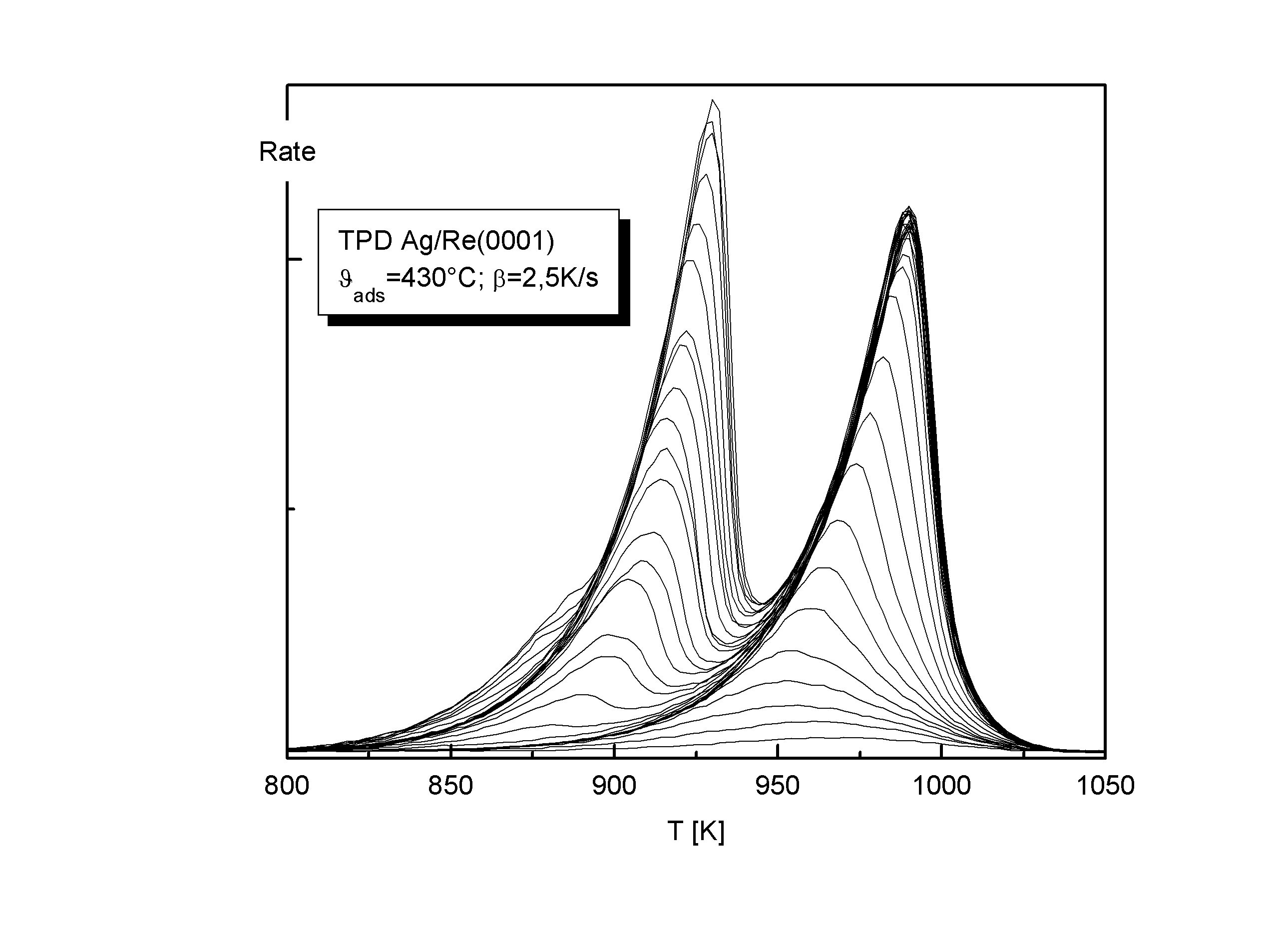
Abbildungen 1, 2
Abb.1 und Abb.2 zeigen Serien von TD-Spektren (R=-dQ /dT=f(T)) zur Untersuchung der ersten beiden Lagen der Systeme Cu/Re(0001) und Ag/Re(0001). Die Spektren zeigen für jeden Zustand eine gemeinsame Niedertemperatur-Anstiegsflanke. Ausnahmen bilden lediglich die Spektren bei sehr kleinen Lagenbedeckungen Q. Der Umstand der gemeinsamen Flankenbildung weist auf eine Desorption nullter Ordnung hin, die immer dann auftritt, wenn sich bei der Desorption die Konzentration der desorbierenden Spezies nicht ändert.
Bei TD-Spektren einer reinen nullten Ordnung ist die gemeinsame Anstiegsflanke bis zum jeweiligen Maximalwert vorhanden, danach sinkt die Desorptionsrate schlagartig auf null ab. Unsere Spektren verlassen jedoch vor Erreichen des Maximalwertes die gemeinsame Flanke zu hohen Temperaturen hin und können daher dort nicht exaktmit einer Kinetik nullter Ordnung beschrieben werden.
Das Abweichen von einer Desorption nullter Ordnung wird bei den Spektren mit kleinen Lagenbedeckungen besonders deutlich. Betrachtet man die Spektren des Systems Cu/Re in einem Bereich bis etwa 0,2ML (s. Abb.3), so erkennt man, daß hier die Maxima der Zustände bei einer identischen Temperatur Tmax liegen, was eine Desorption erster Ordnung nahelegt. (Hierbei ändert sich die Konzentration der desorbierenden Spezies linear mit der Desorptionsrate.)
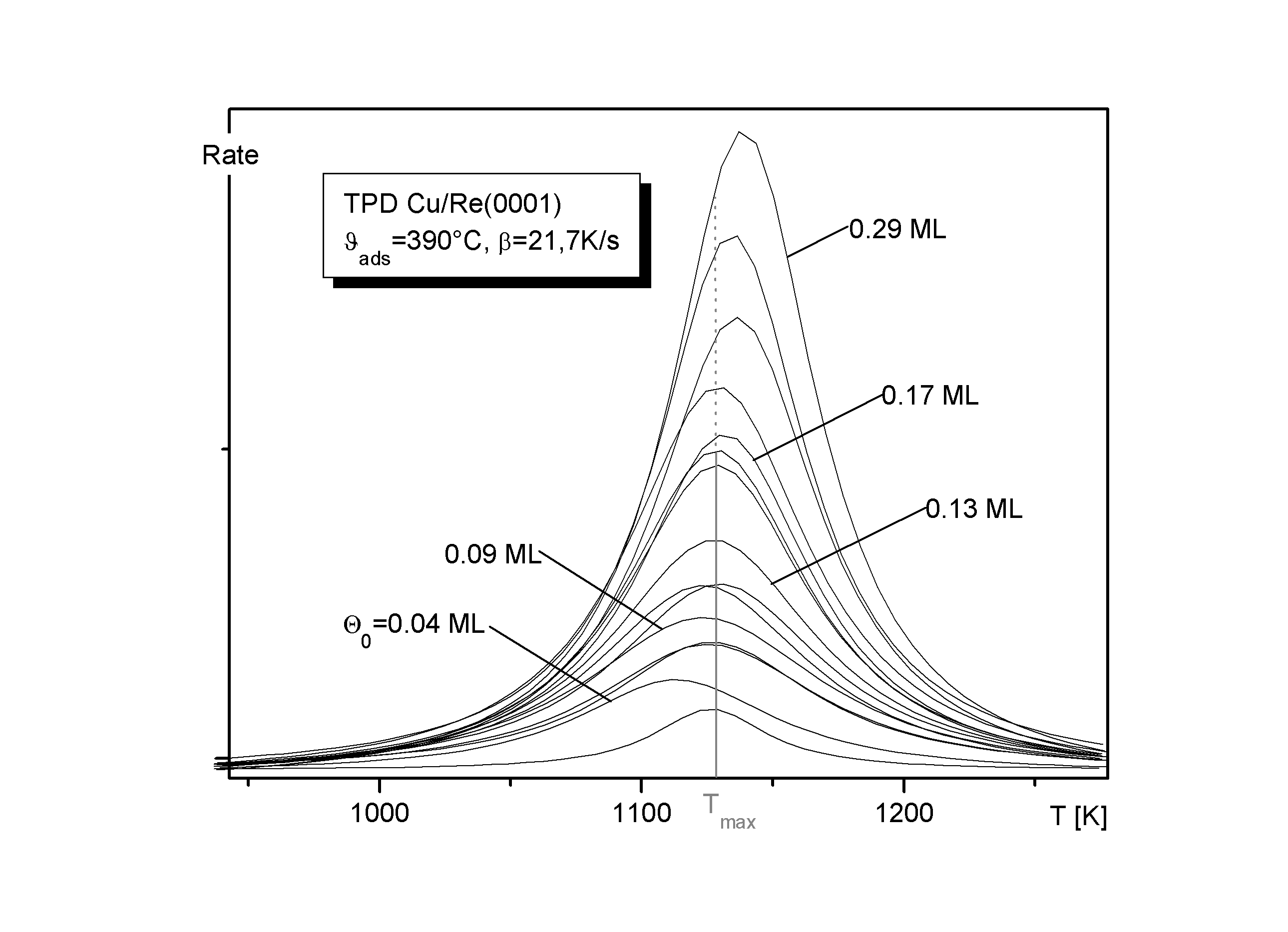
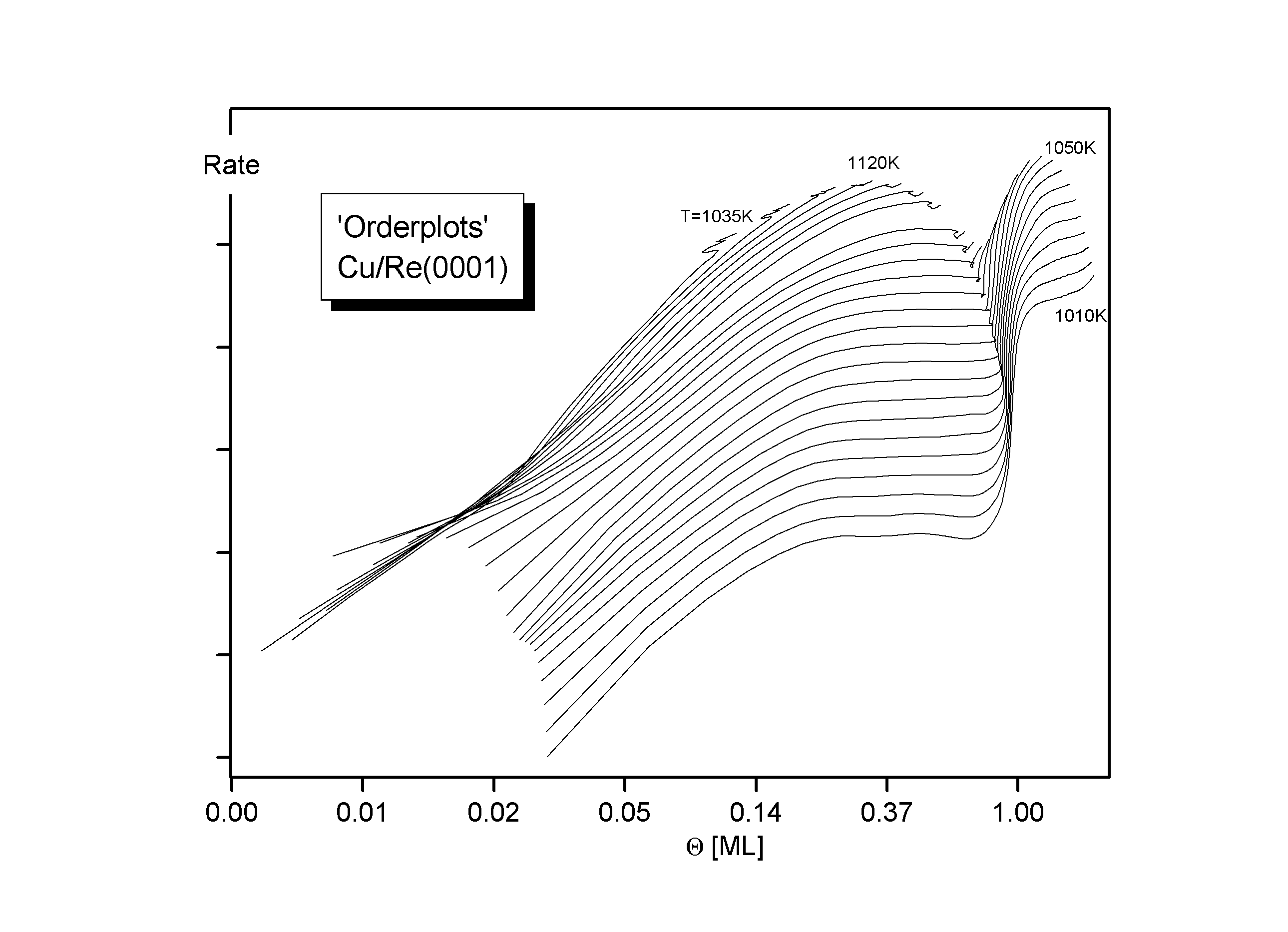
Abbildungen 3, 4
Besonders gut lassen sich die Verhältnisse durch Orderplots verdeutlichen, die eine doppeltlogarithmische Darstellung der Desorptionsrate R in Abhängigkeit vom Bedeckungsgrad Q bei konstanter Temperatur sind (s. Abb.4). Dieser Anstieg der Orderplots entspricht nach
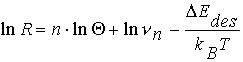 (12)
(12)
und für T=const. der Desorptionsordnung n und soll im folgenden näher beschrieben werden. Die Spektren können in mehrere Q -Bereiche eingeteilt werden, in denen ihre Steigung jeweils konstant n=0 oder n=1 ist:
Abbildungen 5, 6
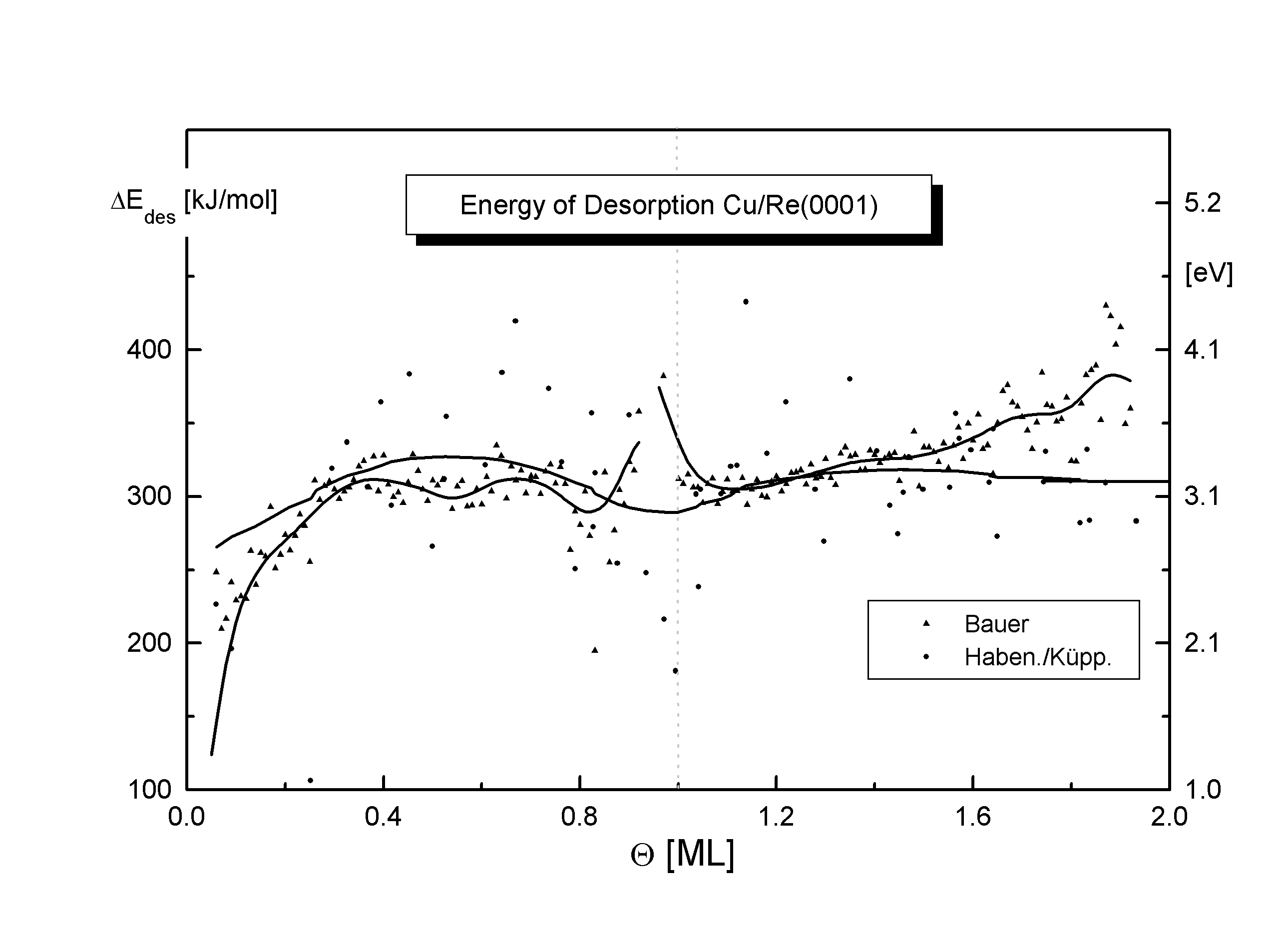
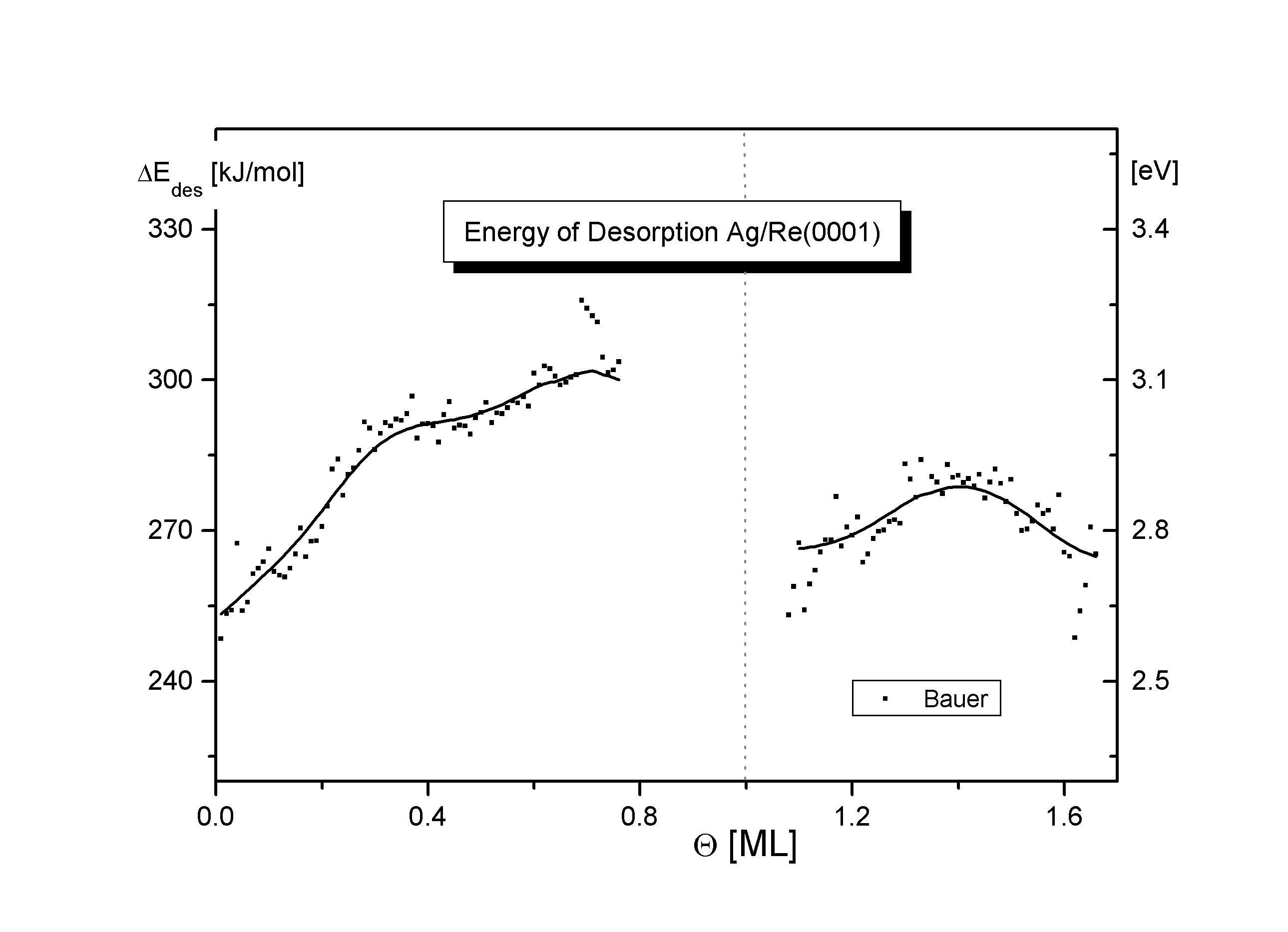
Abb.5 und 6 zeigen den Verlauf der Desorptionsenergie als Funktion des Bedeckungsgrades für die beiden untersuchten Adsorbatsysteme (ermittelt u.a. durch Verfahren nach Bauer [B75/1], Habenschaden/Küppers [HAB83]).
Der Anstieg der Kurven zu Beginn und Ende der ersten Lage ist mit einer Desorption aus einem Einphasengebiet erklärbar, in dem die größere Konzentration der Adteilchen eine verstärkte interne attraktive Wechselwirkung und damit eine steigende Desorptionsenergie bewirkt. Der konstante Verlauf im mittleren Bereich der ersten Lage ist ein weiterer Hinweis auf eine Desorption mit vorgelagertem Gleichgewicht in einem großem Bereich, wobei die Konzentration und damit die Wechselwirkungen der desorbierenden Spezies konstant bleibt. Beim Cu ist der Bereich des konstanten Verlaufs der Desorptionsenergie stärker ausgebildet als beim Silber. Dies ist ein Anzeichen dafür, daß die Desorption der ersten Lage beim Silber mehr aus der reinen Phase abläuft. Der Anstieg von D Edes bis Q » 0,15 (Cu und Ag) setzt sich beim Ag fort. Gittermisfitwechselwirkungen beim Übergang zwischen den Lagen haben den Abfall der Desorptionsenergie zum Ende der ersten Kupferlage und besonders der zweiten Silberlage zur Folge.
Auch AES-Spektren liefern Hinweise auf einen Phasenübergang. In einer Darstellung der Intensitätsverhältnisse von Adsorbat und Substrat sollten sich bei einem Lagenwachstum pro Lage Bereiche mit konstantem Anstieg ausbilden. Für die zweite Lage ist dies annähernd gegeben. Im Bereich der ersten Lage ist der Anstieg der Kurve eher exponentiell, was nach [G90/2] ein Hinweis auf einen Phasenübergang ist.
![]()
Es soll hier eine einfache Möglichkeit vorgestellt werden, um aus den Daten einer Serie von Thermodesorptionsspektren in einem T(Q)-Diagramm die Phasengrenze reine 2D-Gas/Mischphase bis etwa zur kritischen Bedeckung Qc zu erhalten. Am Beispiel des b3-Zustandes des Systems Cu/Re (1. Monolage) soll dies demonstriert werden.
Jedem Spektrum der Serie (mit einer bestimmten Anfangsbedeckung) wird die Grenztemperatur Tg als diejenige Temperatur entnommen, bei der die Spektren die gemeinsame Flanke verlassen (die also den Übergang (2)®(1) kennzeichnet [N84/1, N85/1, N86/1]). Man erhält so als erstes das Wertepaar (Q 0; Tg) (s. Abb.7). Als zweites werden die Spektren von der Hochtemperaturseite her integriert, um den Verlauf des Bedeckungsgrades mit der Temperatur Q =f(T) zu erhalten. Die Serie der integrierten Thermodesorptionsspektren wird dann invers, also in der Form T=f(Q ) in der so erhaltenen Zustandsebene des Systems dargestellt.
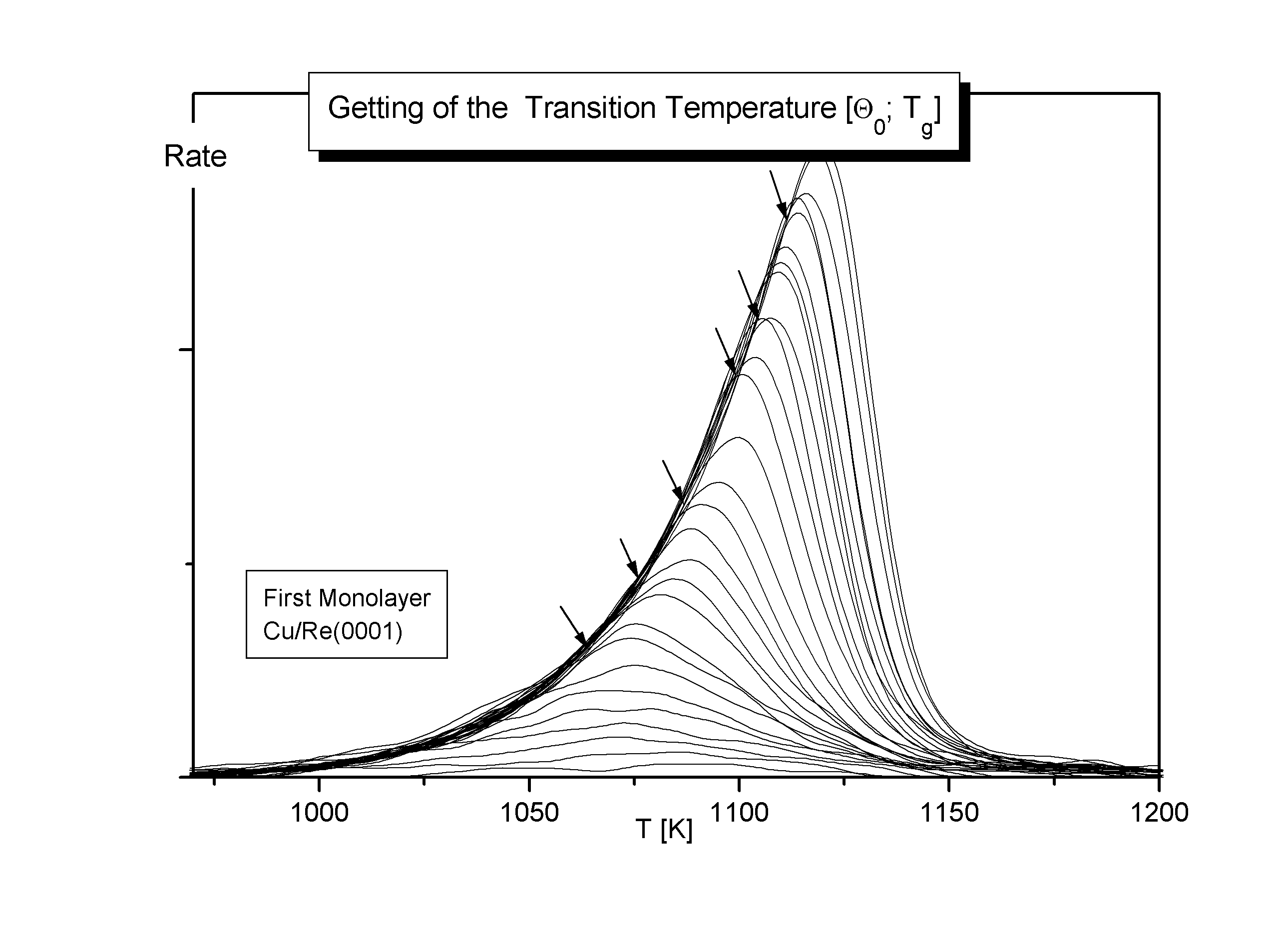
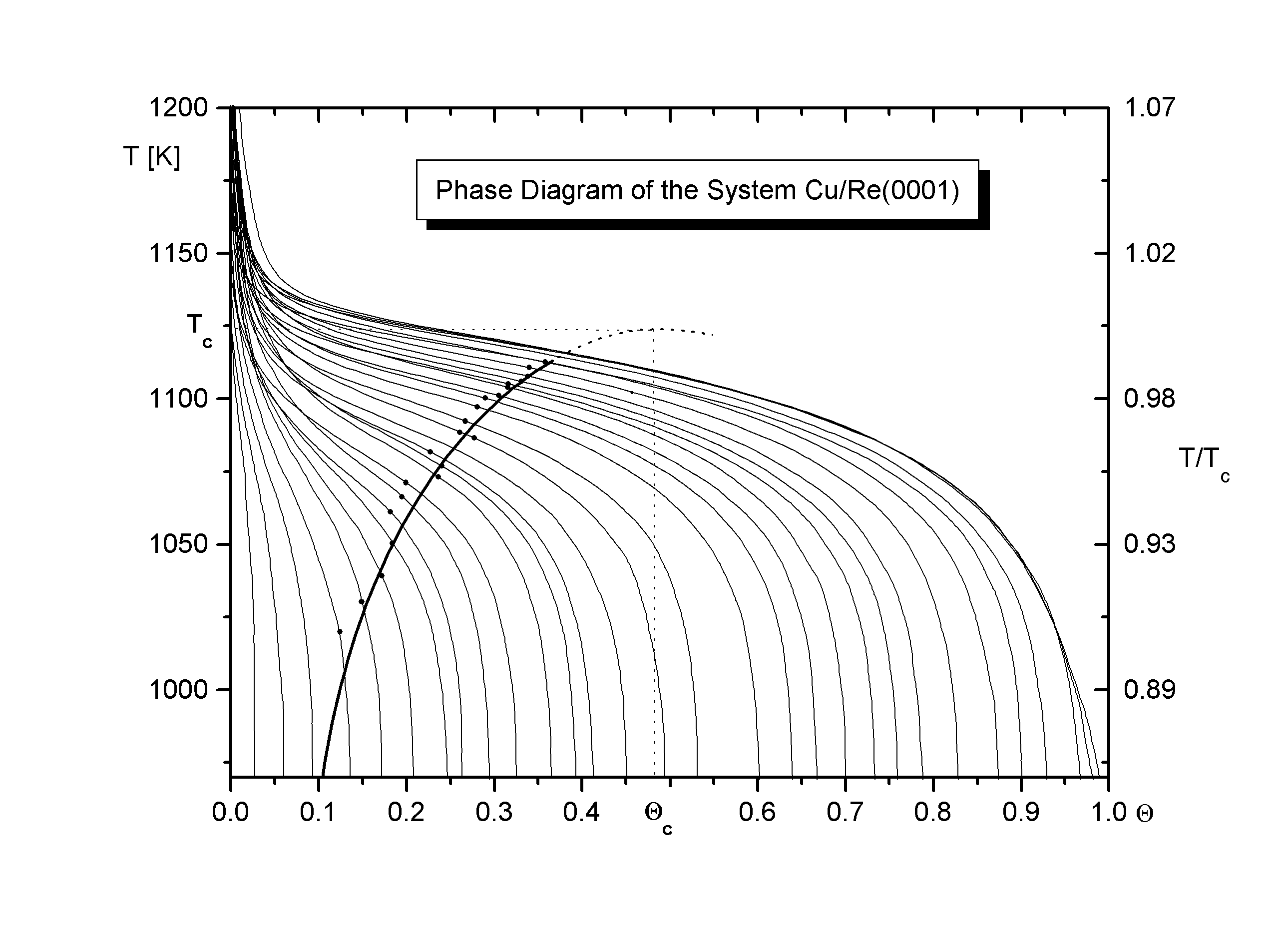
Abbildungen 7, 8
Auf den Desorptionslinien wird jeweils das Wertepaar (Q 0; Tg) eingetragen. Dadurch kann das Wertepaar zum Wertetripel (Q 0; Tg; Q g) erweitert werden, indem man die zu Tg gehörige Phasengrenzbedeckung dem Diagramm entnimmt. Außerdem können die Punkte zu einer Linie verbunden werden, die die Grenze zwischen der reinen Phase und der Mischphase markiert (s. Abb.8). Diese Phasengrenzlinie kann so natürlich nur bis zu dem Punkt bestimmt werden, an dem die Desorptionslinien die Phasengrenze durchschneiden. Um die kritische Temperatur Tc und die kritische Bedeckung Q c zu erhalten, wird berücksichtigt, daß Phasengrenzen für Adsorbatsysteme wie die unseren in den verschiedenen Näherungen (z.B. Bragg-Williams- oder Quasichemische Näherung) zunächst ein Maximum und dann den Punkt (Q =1; T=0) durchlaufen. Ein solches Verhalten wurde auch experimentell für andere Adsorbatsysteme gefunden [B86/2].
Diese Kenntnis erlaubt es, den Kurvenverlauf dementsprechend zu extrapolieren, und man erhält das Maximum der Kurve, dessen Koordinaten die kritischen Werte sind.
Gemäß
(7)
läßt sich aus der kritischen Temperatur die interne Wechselwirkungsenergie bestimmen. In Tab.1 ist diese zusammen mit den kritischen Werten einigen bekannten Literaturdaten gegenübergestellt.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Die kritischen Punkte der Systeme Cu/W und Cu/Mo liegen weit außerhalb des Mischphasengebietes des Systems Cu/Re. Tc ist in den von Bauer vermessenen Systemen größer. Die Wechselwirkungen sind hier stärker, und somit kann sich bei höheren Temperaturen eine Phase ausbilden, die z.T. aus kondensierten Teilchen besteht. Qc ist in diesen Systemen kleinerer. In der BWA wird eine ideal glatte Oberfläche vorausgesetzt. Die geringe Korrugation der (0001)-Oberfläche gegenüber der (110)-Oberfläche bewirkt vermutlich, daß das Cu/Re-System die Vorrassetzungen der BWA besser erfüllt als Cu/Mo bzw. Cu/W, so daß Qc näher bei Q=1/2 liegen sollte.
Die Werte der Systeme Ag/Re und Ag/W stimmen recht gut überein. Die Korrugation der Oberfläche ist vermutlich für die größeren Silberatome nicht so deutlich zu spüren wie für die Kupferatome.
Als ein weiterer Parameter wurde die untere Grenzbedeckung bestimmt. Das ist die Bedeckung, bei der die Desorptionslinien ausschließlich im 2D-Gasphasengebiet liegen, bzw. die Anfangsbedeckung, bis zu der die Thermodesorptionsspektren überhaupt nicht in der gemeinsamen Anstiegsflanke der Spektren zu höheren Anfangsbedeckungen laufen:
Cu/Re: Q g,u»0,1ML (T=950K) Ag/Re: Q g,u» 0,2ML (T=800K)
Anschaulichkeit und Aussagekraft der Daten wird durch die dreidimensionale Darstellung des Desorptionsvorganges verbessert. Dazu wird die Desorptionsrate R als Funktion der Zustandsebene des Systems (Q ; T) dargestellt [W98/1]. Man erhält so Desorptionspfade auf der Desorptionsfläche, die für den b 3-Zustand (1.ML) des Systems Cu/Re in Abb.9 dargestellt sind. Als Projektion in die (T; R)-Ebene ergeben sich die durch das TD-Experiment zugänglichen TD-Spektren, eine Projektion in die (Q; R)-Ebene liefert die Layerplots. In der (Q ; T)-Zustandsebene sind die integrierten TD-Spektren zu erkennen. Diese Ebene ist gleichzeitig das Phasengebiet. Der Verlauf der Phasengrenze ist in die TD-Fläche sowie in die drei Projektionsebenen eingezeichnet. Die Projektion in die (Q ; T)-Ebene ist die eigentliche Phasengrenze. Die Entnahme der Meßpunkte der Phasengrenze ist in der (T; R)-Projektion ersichtlich. In der (Q ; R)-Ebene erhält man Layerplots [W98/1, M92/1] Die Desorption vollzieht sich in dieser Darstellung von rechts nach links. Dem Diagramm kann die Desorptionsrate zum momentanen Bedeckungsgrad, ausgehend von einer bestimmten Anfangsbedeckung, entnommen werden.
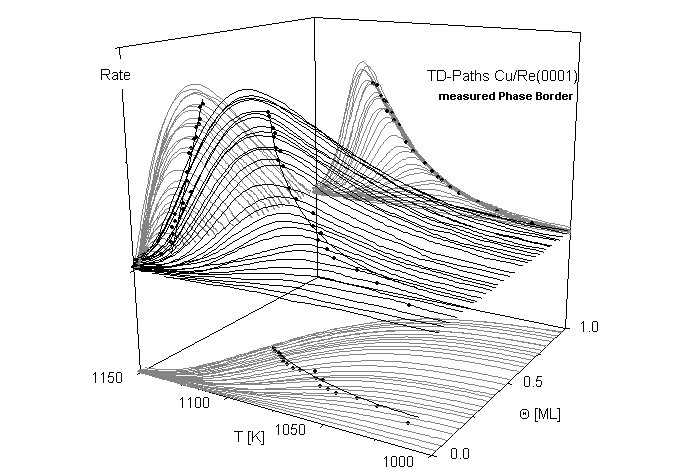
Abbildung 9
In einem weiten anfänglichen Bereich zeigen fast alle Desorptionslinien (überwie-gend diejenigen mit mitt-leren bis großen Bedeckungen) einen konstanten Anstieg. Dieser Effekt ist damit zu erklären, daß die exponentielle Abhängigkeit der Desorptionsrate (für n=0) mit der Temperatur hier durch die besondere Art der Projektion nicht sichtbar ist. D.h., das Verlassen der gemeinsamen Anstiegsflanke in den TD-Spektren dokumentiert sich hier durch das Abweichen vom linearen Verlauf.
Der Verlauf der Phasengrenze erscheint in einem weiten Bereich, nämlich zwischen 0,17 und 0,35 (Cu), bzw. 0.22 und 0,35 (Ag) linear. Auch hier ist durch die besondere Auftragung der eigentlich exponentielle Anstieg der Phasengrenzbedeckung Q g zu einer linearen Funktion gestreckt worden. Der konstante Anstieg der Phasengrenze kennzeichnet indirekt die Temperaturabhängigkeit von Qg und damit auch die des Phasengleichgewichtes zwischen der reinen Phase und der Mischphase. Die Grenzbedeckung Q g, die aus Abb.8 nur mit geringer Genauigkeit entnommen werden konnte, ist hier sehr einfach zu bestimmen. Extrapoliert man den linearen Teil der Phasengrenze auf die Q -Achse, erhält man mit großer Genauigkeit den Wert von Q g, vgl. auch Abb.11 und Abb.12. Es ergibt sich:
Cu/Re: Q g,u=0,16ML Ag/Re: Q g,u=0,22ML.
Werden in Abb.9 die Punkte gleicher Temperatur bzw. gleichen Bedeckungsgrades verbunden, so erhält man die Desorptionsisothermen bzw. Desorptionsisosteren. In Abb.10 ist die Desorptionsfläche durch die Isothermen und Isosteren gegeben, deren Projektionen in die entsprechenden Ebenen ebenfalls dargestellt sind [W98/1].
Die gemessene Phasengrenze (Flankenmethode) ist auch hier eingezeichnet, ebenso in der (Q ; T)-Ebene die mit Tc(mess) simulierte (BWA-) Phasengrenze. Der Mischphasenbereich des Systems Cu/Re und besonders auch Ag/Re wird durch die (BWA-) Simulation unterrepräsentiert. Dies beruht darauf, daß sich die Systeme nicht vollkommen ideal verhalten. Abweichungen werden insbesondere durch Mehrkörperwechselwirkungen und Fehlstellen des Gitters bedingt. Beim Silber muß der positive Gittermisfit berücksichtigt werden, der eine stärkere repulsive Wechselwirkung der Teilchen hervoruft. Die simulierte (BWA-) Phasengrenze wurde von hier in die TD-Fläche sowie in die (Q ; R)- und die (T; R)-Ebenen projeziert.
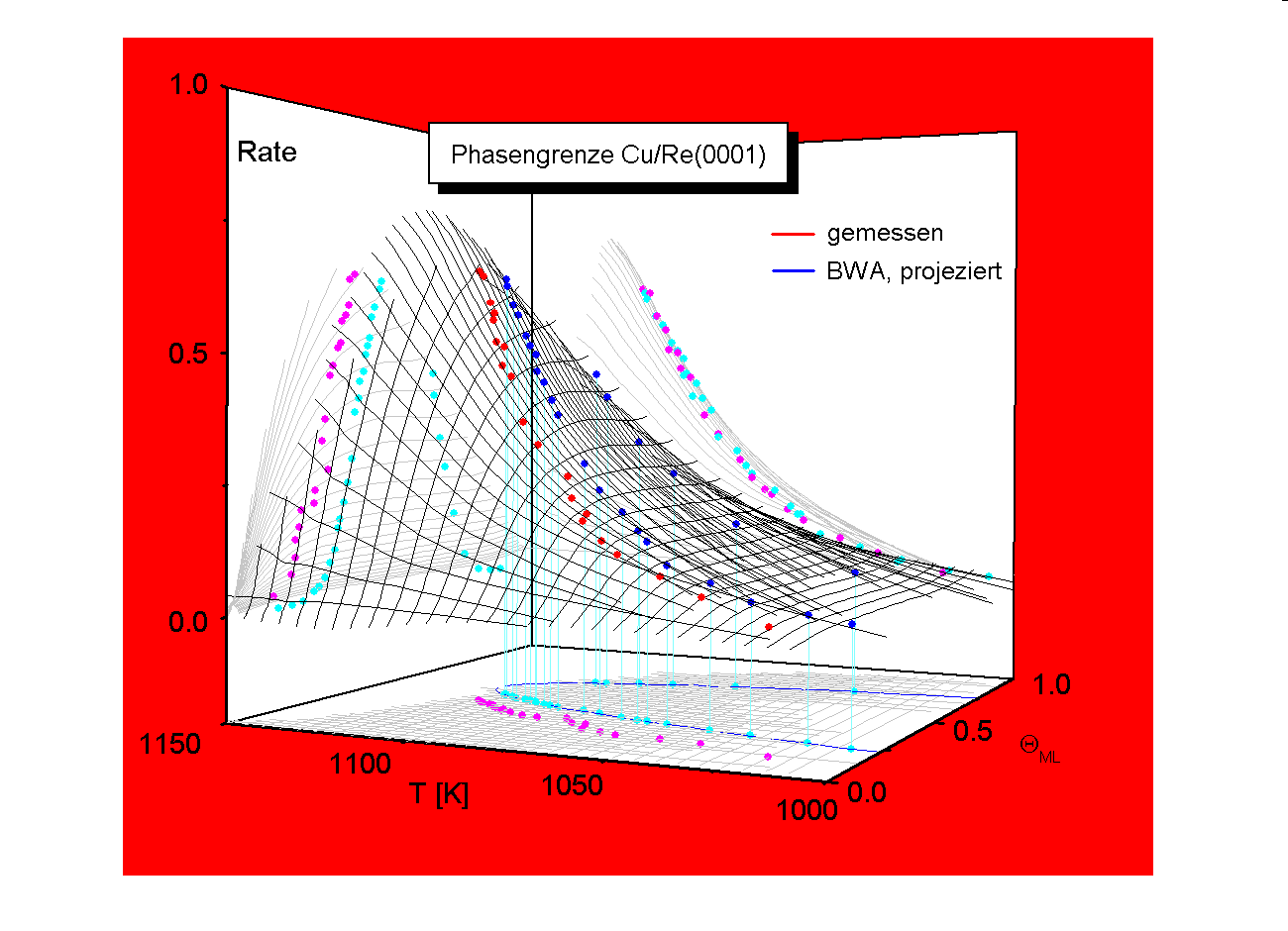
Abbildung 10
Der Verlauf der (Q ; R)-Projektion der BWA-Phasengrenze ist in einem großen Teil des ansteigenden Astes ebenso linear wie der der gemessenen Grenzlinie. Auch aus den Isosteren kann sehr genau Q g,u entnommen werden. Es ist der Parameter der Isostere, die als erste den gemeinsamen Verlauf der Isosteren höherer Bedeckungen verläßt.
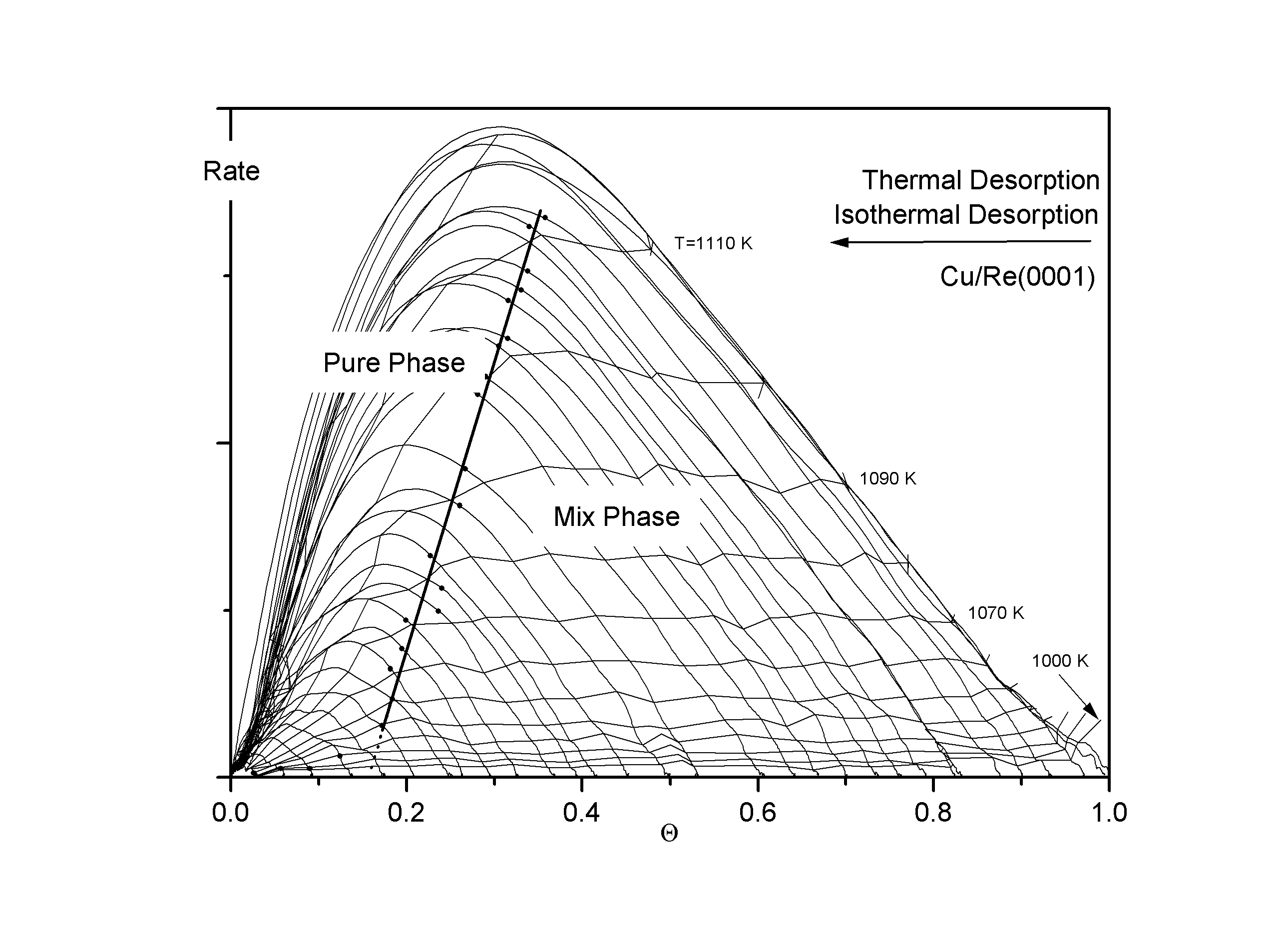
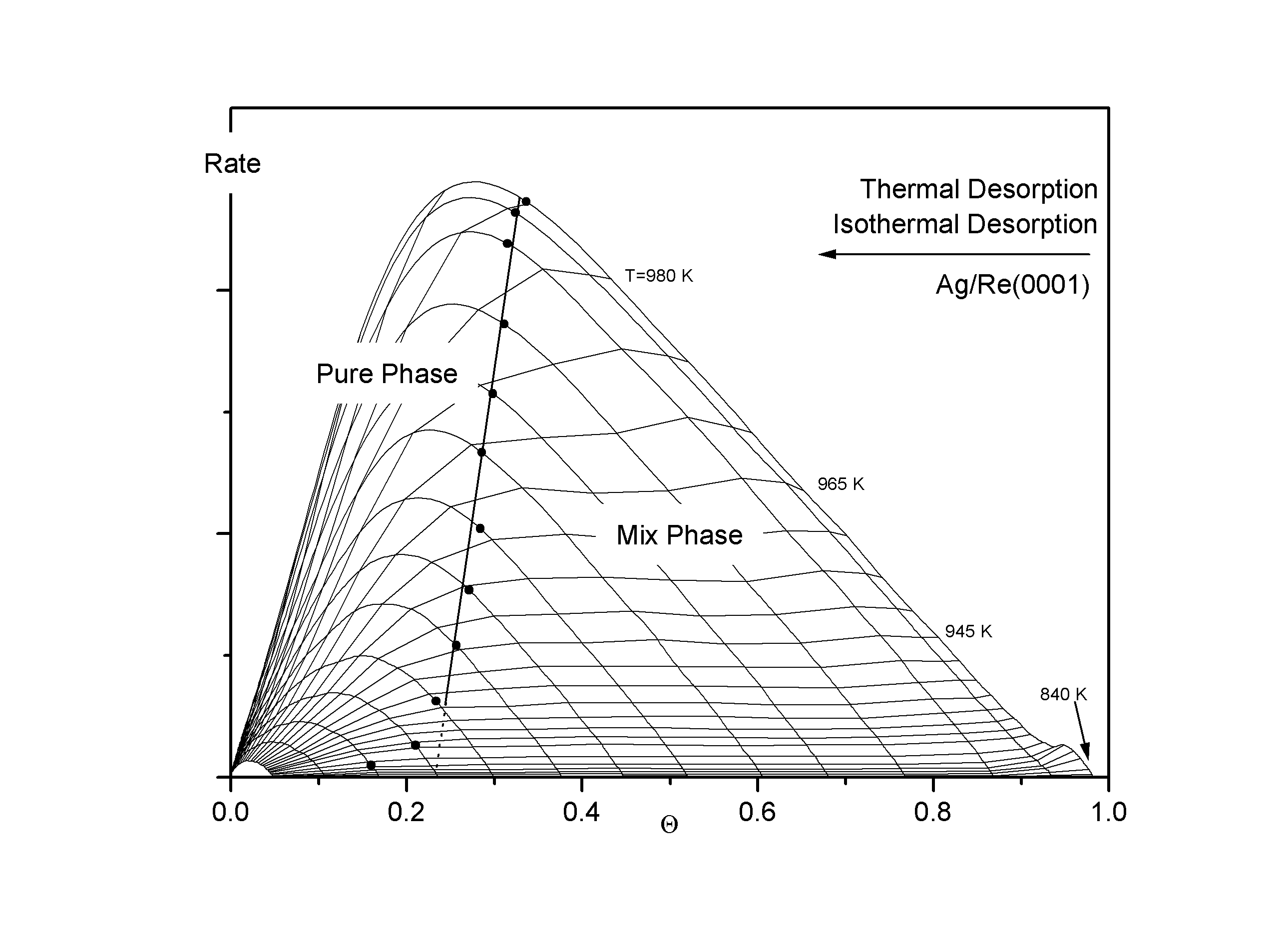
Abbildungen 11, 12
In den Abb.11 und 12 sind sowohl die Desorptionslinien (Layerplots), als auch die Desorptionsisothermen dargestellt. Hier wird die gute Übereinstimmung des Verlaufes der mittels der Flankenmethode gewonnenen Phasengrenze für den Übergang ins Mischphasengebiet (1)® (2) mit den Knicken der Isothermen deutlich.
Für den Übergang (2)® (1) lassen sich leider keine genauen Aussagen treffen, da dieser Bereich durch das Desorptionsexperiment nicht erfaßt wird. Allerdings sollte der Übergang außerhalb des Desorptionsbereiches liegen, da kein erneuter Anstieg der Isothermen zu verzeichnen ist. (Lediglich ab 0.9 ML kann ein geringer Anstieg vermutet werden.)
![]()
Die Thermodesorptionsspektren der Systeme Cu/Re(0001) und Ag/Re(0001 insbesondere für die erste aber auch für die zweite Lage sind in einem großen Bereich durch den Verlauf in einer gemeinsamen Anstiegsflanke gekennzeichnet. Dieses Verhalten kann annähernd mit einer Desorptionsordnung von n=0 beschrieben werden.. Die Spektren für kleine Anfangsbedeckungen zeigen jedoch ein davon abweichendes Verhalten. Ihre Maxima liegen bei gleicher Temperatur, ihre Desorptionsordnung ist damit n=1. Dies läßt den Schluß zu, daß es während der Desorption zu einem Wechsel der Desorptionsordnung kommt. Dieser Umstand spiegelt sich auch in der Form der TD-Spektren zu mittleren bis hohen Lagenbedeckungen wider. Diese Spektren weichen kurz vor dem Erreichen des Maximalwertes von der gemeinsamen Anstiegsflanke ab. (Spektren reiner nullter Desorptionsordnung würden in einer gemeinsamen Flanke ansteigen, um senkrecht auf null abzufallen, wenn Q =0 wird.) Die Desorption vollzieht sich also bei kleinen Bedeckungsgraden und hohen Temperaturen nach erster, sonst nach nullter Desorptionsordnung.
Dieser Befund wird auch in den Orderplots, einer doppeltlogarithmischen Darstellung berrechnetrer Desorptionsisothermen gut wiedergegeben. Bis zu einem Bedeckungsgrad von Q » 0.2 ist n=1, danach n=0.
Innerhalb der TD-Fläche (R=f(Q ; T)) ist der Bereich mit n=0 durch einen in T-Richtung exponentiellen, in Q -Richtung jedoch geraden Verlauf gekennzeichnet. Ebenfalls gerade ist der Verlauf der Layerplots (der Projektion der TD-Pfade in die (Q ; R)-Ebene).
Der Verlauf der Desorptionsenergie kann ebenfalls in mehrere Bedeckungsgradgebiete eingeteilt werden, die durch ein bestimmtes Verhalten gekennzeichnet sind.
Bei kleinen Bedeckungsgraden bildet sich auf der Oberfläche eine zweidimensionale Gasphase aus. Diese besteht aus Teilchen, die sich auf der Oberfläche frei bewegen können. Bei höheren Bedeckungen ( Cu: Q gu=0.16, Ag: Q gu=0,22) kommt es zu starken Wechselwirkungen zwischen den Adteilchen, was dazu führt, daß ein bestimmter Anteil kondensiert. Diese kondensierte Phase steht mit der 2D-Gasphase im Gleichgewicht.
Je nach dem, wie groß der Bedeckungsgrad beim momentanen Stand der Desorption ist, liegen unterschiedliche Verhältnisse auf der Oberfläche vor, und es sind verschiedene Desorptionskinetiken beobachtbar. Wird aus der reinen (2D-Gas-) Phase desorbiert, ist eine Desorptionsordnung von n=1 zu beobachten, die die Konzentrationsabhängigkeit des Vorgangs dokumentiert. Liegt der Fall der Koexistenz von 2D-Gas- und Kondensatteilchen auf der Oberfläche vor, desorbieren auch nur 2D-Gasteilchen, deren Konzentration allerdings durch ein vorgelagertes Gleichgewicht hoher Reaktionsgeschwindigkeit (C® G) konstant gehalten wird. Dabei ist die Desorptionsordnung null.
Die Gültigkeit der Polanyi-Wigner-Gleichung ist hier allerdings eingeschränkt. Für die Desorptionskinetik bestehen, je nach momentanem Bedeckungsgrad, zwei Möglichkeiten, entweder bedeckungsgradabhängig oder nicht. Um die Desorption vollständig beschreiben zu können, sind zwei Ratengleichungen (z.B. 10,11) nötig.
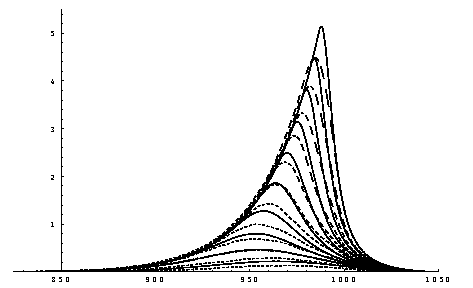
Abbildung 13 gemessene (gestrichen) und simulierte (durchgezogen) TD-Spektren für die erste Lage des Systems Ag/Re(0001)
Dies wird in Abb. 13 deutlich. Hier sind die experimentellen TD-Spektren für die Desorption aus der ersten Lage simulierten Spektren gegenübergestellt. Der Simulation liegen Ratengleichungen der Form (11, 12) zugrunde, zwischen denen mittels einer Umschaltfunktion bei Q gu gewechselt wird. Die Parameter sind die o.a. Meßwerte. Es ist hierbei zu beachten, daß absichtlich D Q 0,mess=0,07, aber D Q 0,rech=0,1 gilt, sodaß simulierten und gemessenen Spektren nicht zur Deckung kommen. Trotzdem ist eine sehr gute Übereinstimmung gegeben.
Durch die Anwendung der Flanken- oder der Isothermenmethode ist es möglich, den Verlauf der Phasengrenze im Desoptionsbereich zu bestimmen. Daraus können die kritischen Werte entnommen werden. Beim Kupfer sind dies Q c=0,48ML, Tc=1120K und beim Silber Q c=0,33ML, Tc=986K. Daraus lassen sich die internen Wechselwirkungsenergien berechnen, nämlich EWW=6,2kJ/mol für das Kupfer und EWW=5,5kJ/mol für das Silber.
Mit Hilfe dieser Energie EWW kann der Verlauf der Desorptionsenergie beschrieben werden. Diese stellt eine Meßgröße dar, die sich aus einem Anteil vertikal (Vz(Cu)=291,4kJ/mol, Vz(Ag)=276,5kJ/mol) und horizontal (EWW) zur Oberfläche zusammensetzt, der einen Anteil von je 2% an der mittleren Desorptionsenergie hat. (D Edes(Cu)=310kJ/mol [W98/1], D Edes(Ag)=293kJ/mol [S98/1]) Der Bereich der Desorption aus dem Einphasengebiet ist dabei durch einen Anstieg gekennzeichnet. Bei der Desorption aus dem Mischphasengebiet ist die Desorptionsenergie konstant. Die Meßwerte stimmen mit Literaturwerten ähnlicher Systeme etwa überein, geringe Abweichungen resultieren aus den unterschiedlichen Oberflächenrauhheiten und Adsorbat/Substrat-Größenverhältnissen.
![]()
B75/1 E. Bauer, H. Poppa, G. Todd, F. Bonczek, "Thermal Desorption of Metals from Tungsten Single Crystal Surfaces", Surf. Sci., 53 (1975) 87.
B85/1 J. Kolaczkiewicz, E. Bauer, "The Law of Corrosponding States for Chemisorbed Layers with Attractive Lateral Interactions", Surf. Sci., 151 (1985) 333.
B86/2 J. Kolaczkiewicz, E. Bauer, "Thermal Desorption Spectroscopy of Ni, Cu, Ag and Au from W(110)", Surf. Sci., 175 (1986) 508.
B87/1 M. Paunov, E. Bauer, "An Adsorption-Desorption Study of Cu on Mo(110)", Appl. Phys. A, 44 (1984) 201.
B88/1 A. Pavlovska, H. Steffen, E. Bauer, "A Comparison of Thermal Desorption Techniques at High Temperatures: Au on Mo(110)", Surf. Sci., 195 (1988) 207.
EST86 P. J. Estrup, E. F. Green, M. J. Cardillo, J. C. Tully, "Influence of Surface Phase Transitions on Desorption Kinetics: The Compensation Effect", J. Phys. Chem., 90 (1986) 4099.
G90/2 He, J. W., Goodman, D. W., "Copper Overlayers on Rhenium (0001)", J. Phys. Chem., 94 (1990) 1496.
K88/1 S. H., Payne, "Desorption from a Two-Phase Adsorbate: Zero or Fractional Order", Surf. Sci., 200 (1988) L433.
K89/1 S. H., Payne, H. J., Kreuzer, "Analyses of Thermal Desorption Data", Surf. Sci., 222 (1989) 404.
K90/1 H. J., Kreuzer, "Theory of Surface Processes", Appl. Phys. A, 51 (1990) 491.
K90/2 H. J., Kreuzer, "Theory of Surface Processes", Surf. Sci., 231 (1990) 213.
K91/3 H. J., Kreuzer in: in: "Dynamics of Gas-Surface Interactions", Eds.: M. N. R. Ashfold, C. T. Rettner, Ch. 6: "Thermal Desorption Kinetics", Royal Soc. of Chem., Cambr. (1991), S. 220 ff..
K95/2 S. H., Payne, H. J., Kreuzer, "Multilayer Adsorption and Desorption", Surf. Sci., 338 (1995) 261.
M92/1 Schlichting, H., Menzel, D., "High Resolution, Wide Range, Thermal Desorption Spectroscopy of Rare Gas Layers: Sticking, Desorption Kinetics, Layer Growth, Phase Transitions and Exchange Processes", Surf. Sci., 176 (1992) 27.
N84/1 K. Nagai, T. Shibanuba, M. Hashimoto, "Zero-Order Desorption Kinetics Based on Phase Equillibrium", Surf. Sci., 145 (1984) L459.
N85/1 K. Nagai, "Rate Expression Incorporating Interaction between Reactants: Application to the Zero Order Desorption Spectra", Phys. Rev. Lett., 54 (1985) 2159.
N86/1 K. Nagai, "A Simple Rate Equation Useful for Adsorptions Systems: Analyses of Thermal Desorption Spectra", Surf. Sci. 176 (1986) 193.
N87/1 K. Nagai, A. Hirashima, "Zero-Order Desorption is Always Observed in Phase Equilibrium within Adsorbates?", Surf. Sci., 187 (1987) L616.
N88/1 K. Nagai, A. Hirashima, "Zero-Order Desorption Kinetics Observed in Phase Coexistence Regions in Adsorbates", Appl.Surf. Sci., 33/34 (1988) 335.
S98/1 Schlatterbeck, D., Parschau, M., Christmann, K., "Silver-Films Grown on a Re(0001)Surface: A Combinated TDS, XPS and D j Study", Surf. Sci., eingereicht.
SAF94 S. A. Safran, "Statistical Thermodynamics of Surfaces, Interfaces and Membranes", Ch. 1.4: "Phase Seperation in Binary Mixtures", Addison-Wesley Publishing Company (1994), S. 21 ff..
SEE88 E. G. Seebauer, A. C. F. Kong, L. D. Schmidt, "The Coverage Dependence of the Pre-exponential Factor for Desorption", Surf. Sci., 193 (1988) 417.
SPA85 M. J. Spaarney, "Thermodynamics (With an Emphasis on Surface Problems)", Surf. Sci. Rep., 4 (1985) 101.
W98/1 Wagner, R., Schlatterbeck, D., Christmann, K., "TD-Spectroscopy of Cu/Re(0001)", in Vorbereitung.
Z81/1 V. P., Zhdanov, "Lattice Gas Model for Description of the Adsorbed Molecules of Two Kinds", Surf. Sci., 111 (1981) 63.
Z85/2 V. P., Zhdanov, "Simple Model for an Adsorbate-Induced Phase Transition", Surf. Sci., 164 (1985) L807.
Z91/1 V. P., Zhdanov, "Arrhenius Parameters for Rate Processes on Solid Surfaces"Surf. Sci. Rep., 12 (1991) 185.