|
|
|
Kapitel A3 | Abschnitt A - Theoretische Grundlagen |
|
|
|
Kapitel A5 | Kapitel A4 - Thermodesorptionsspektroskopie |
|
|
|
Kapitel A3 | Abschnitt A - Theoretische Grundlagen |
|
|
|
Kapitel A5 | Kapitel A4 - Thermodesorptionsspektroskopie |
Seit die ersten Messungen zur temperaturprogrammierten Desorption von Apker im Jahre 1948 durchgeführt worden sind, hat sich in bezug auf die Meßpraktik aber auch im Hinblick auf die Auswertungsmethodik viel geändert. Der Einsatz der Computertechnik hat dazu einen großen Beitrag geleistet.
Thermodesorptionsspektroskopie (TDS) stellt ein gutes Werkzeug dar, um energetische und kinetische Prozesse auf Oberflächen zu untersuchen. So ist es möglich, Aussagen über Wachstumsmodi adsorbierter Spezies zu treffen, die Bindungsenergie von Adteilchen auf der Oberfläche zu untersuchen und Informationen über die Wechselwirkungen der Adteilchen untereinander zu erhalten.
Erhält ein Adteilchen genügend Energie, so kann es desorbieren, vgl.
|
|
(A63) |
Auf Grund der Erwärmung des gesamten Systems (der Probe) desorbieren die Teilchen, und zwar in der Reihenfolge ihrer Bindungsenergie, also die am schwächsten gebundenen zuerst. (Der Desorption können weitere Prozesse vorgelagert sein, wie etwa Dissoziationen von Adteilchen, Phasenübergänge, Übergänge zwischen verschiedenen Bindungszuständen, usw.) Dabei bilden sich ein oder mehrere Desorptionsmaxima bei den dazu gehörigen Temperaturen Tmax aus.
Die desorbierten Teilchen werden in einem Massenspektrometer detektiert und man erhält Spektren der Form Ii (µ pi) = �(T). Wenn das Reaktionssystem schnell genug gepumpt wird, gilt die proportionale Abhängigkeit
Durch Integration erhält man die Bedeckung zur entsprechenden Zeit bzw. Temperatur.
|
|
(A64) |
Der einfachste Fall eines TD-Spektrums ist die Ausbildung eines einzigen Zustandes, wie z. B. bei der Sublimation. Oftmals treten Spektren mit mehreren Zuständen (Multipeakspektren) auf, die man z. B. einzelnen Adsorbatlagen zuordnen kann. Diese lassen sich auf Grund starker Überlappung der einzelnen Zustände z. T. nur schwer separieren.
Dies resultiert einerseits aus der gleichzeitigen Desorption mehrerer unabhängiger Zustände mit einer damit verbundenen Linearkombination individueller Peaks i, andererseits aber auch daraus, daß das System unter verschiedenen Bedingungen (Variation von Q, b, T) spektroskopiert wird und die Zustände miteinander wechselwirken.
Je nach Bindungsplatzenergetik kann man so auch für Spektren, die zu einer Lage gehören, mehrere TD-Zustände erhalten. Die Aufspaltung der einzelnen Zustände vergrößert sich mit der Erhöhung des Unterschiedes der Wechselwirkungen der desorbierenden Spezies [Ada74/1]. Besonders leicht tritt eine solche Aufspaltung bei repulsiven Wechselwirkungen auf [PWK93/1], und zwar ab Bedeckungsgraden, bei denen die Teilchen direkte Nachbarn auf den nächsten Adsorptionsplätzen besitzen.
In mehrdeutigen Fällen muß hier auf die Monolageneichung durch andere Meßmethoden, z. B. Augerelektronenspektroskopie, zurückgegriffen werden.
Die totale Desorptionsrate, die man aus den experimentellen TDS erhält, ist gegeben durch:
|
|
(A65) |
bzw. unter Verwendung der Lebensdauer formuliert:
|
|
(A66) |
Bei sehr guter Aufspaltung der Zustände kann man diese durch einfache Subtraktion trennen. Eine weitere Möglichkeit der Separation besteht darin, die Fläche für den ersten Desorptionszustand aufzuintegrieren und damit alle weiteren Kurven zu simulieren. In anderen Fällen muß auf eine Trennung verzichtet oder versucht werden, alle Prozesse, die zu der Überlappung führen, zu berücksichtigen [PLM02/1].
Bisher wurden zahlreiche Methoden entwickelt, um all diese Informationen aus den TD- Spektren zu extrahieren. Bereits in der Vergangenheit erschienen hierzu gute Übersichtsartikel z. B. von de Jong et al. [JoN90/1], Yates et al. [MSG87/1] oder King [Kin75/1].
Eine andere Möglichkeit, aus TD-Spektren Informationen zu erhalten ist, solche Spektren durch den Einsatz von Näherungen zu simulieren und die so erhaltenen gerechneten Spektren durch die Wahl geeigneter Parameter den Meßwerten anzupassen. Umfangreiche Arbeiten dieser Art stammen u. a. von Kreuzer [PaK95/1, PKP96/1, PKK96/1], Zhdanov [Zhd83/1], oder Nagai [Nag86/1].
All die in der Literatur vorgestellten Methoden zur Informationsgewinnung aus dem TD-Experiment stellen für sich allein gute und interessante Wege dar, um die gewünschten Aussagen zu erhalten. Sie sind zum Teil sehr unterschiedlich, was die Form der Darstellung, aber auch die verwendete mathematische Näherung, die zur Auswertung benutzt wird, betrifft. Nur durch eine Betrachtung aller gewinnbaren Informationen läßt sich deren Vielzahl ordnen, und nur dann lassen sich Meß- und Systemparameter mit guter Genauigkeit extrahieren.

| |
| Abbildung A13 TD-Spektren des Systems Cu/Re(0001) [WSC99/1] b = 7,7 K/s. |
|
Der große Vorteil des TD-Experiments liegt in seiner einfachen Anwendbarkeit, die mit der Möglichkeit gekoppelt ist, zugleich thermodynamische und kinetische Parameter zu erhalten. Der Wirkungsgrad des Experiments beträgt 100%, d. h., es wird tatsächlich nur der Desorptionspfad aktiviert. Dies geschieht durch die Zuführung einer bestimmten Wärme Qdes. Als den Zustand des Systems charakterisierende Größe wird die Temperatur in Form einer linearen Heizrampe vorgegeben und kontrolliert. Als das System selbst beschreibende Größe wird die Desorptionsrate (Intensität des Massenspektrometersignals der betrachteten Adteilchenart) gemessen. Insofern ist die Messung selbst zweidimensional. Man erhält Spektren der Form
Schon hieraus lassen sich einige Aussagen über das System gewinnen. Aus der Form der TD- Spektren läßt sich bereits die Desorptionsordnung abschätzen. Besitzen die Spektren eine gemeinsame Anstiegsflanke wie in
Aus der Lage der Peakmaxima läßt sich nach Redhead [Red62/1] ungefähr die Bindungsenergie der Adteilchen an die Oberfläche abschätzen. Dabei wird z. B. für Desorption nach erster Ordnung ein linearer Zusammenhang zwischen der Temperatur des Peakmaximums und der Desorptionsenergie abgeleitet (was sicherlich nur eine sehr grobe Näherung darstellt und für ganz einfache Systeme praktikabel ist [NMW88/1]).
Neben der Temperatur ist der momentane Bedeckungsgrad Q die zweite wichtige Kenngröße, die den Zustand des Systems beschreibt. Q stellt sich im TD-Experiment entsprechend der Systemeigenschaften ein und wird zunächst bei der Messung nicht berücksichtigt. Glücklicherweise ist es bei der Auswertung der TDS jedoch möglich, Q als relative Größe nachträglich zu berechnen. Dazu wird in Betracht gezogen, daß die gemessene Rate natürlich der differentiellen Änderung des Bedeckungsgrades entspricht.
|
|
(A67) |
Durch die numerische Integration der Werte der Desorptionsrate von der Hochtemperaturseite bis zur jeweiligen Temperatur Ti, also entgegengesetzt der Desorptionsrichtung, können Wertepaare (Qi; Ti) gewonnen werden.
|
|
(A68) |
(NML ... Monolagen-Normierfaktor)
Man erhält so temperaturabhängige Bedeckungsgradkurven. Diese Kurven (s.
Die gemessene Desorptionsrate R ist von zwei Variablen, nämlich Bedeckungsgrad und Temperatur, abhängig.
|
|
(A69) |

| |
| Abbildung A14 Zustandsebene des System Cu/Re(0001) mit TD-Trajektorien [WaC00/1]. |
|
Gleichung (A69) (n
... Präexponentialfaktor, n ... Desorptionsordnung, R ... Gaskonstante, Qdes ... zugeführte Wärme) ist eine Verallgemeinerung der Polanyi-Wigner-Gleichung und beschreibt den TD-Vorgang vollständig, solange es sich um nur eine desorbierende Spezies auf identischen Adsorptionsplätzen handelt und nur ein Elementarschritt geschwindigkeitsbestimmend ist. Je nach beschriebenem System können die Größen in
Qdes wird oftmals durch die Desorptionsenergie -Edes ausgedrückt, die dem System in Form von Wärme zugeführt wird. Diese Notation soll auch hier angewendet werden.
In Gleichung (A69) können sowohl entropische als auch enthalpische Effekte eingehen, deren gegenseitige Wechselwirkungen gerade die Abhängigkeiten von Q und T hervorrufen können. Weiterhin kann es sich bei der Desorption um einen aktivierten oder nicht aktivierten, assoziativen oder dissoziativen Vorgang handeln. Auch das Auftreten eines Precursor-Zustandes kann in die Gleichung eingehen. Ferner stellt sich die Frage, ob sich das System während der Desorption im thermodynamischen Gleichgewicht befindet, oder ob auf Grund der Anwendung sehr hoher Heizraten beispielsweise kinetische Hemmungen auftreten.
Durch DF
-Messungen am System Ag/Ru(0001) im Bereich von 400 K ... 900 K konnten Wandelt et al. keinen Unterschied bei Heizraten von 1 K/s und 10 K/s erkennen [NSW95/1]. Dies ist ein deutliches Zeichen dafür, daß sich das System bei nicht zu hohen Heizraten im thermischen Gleichgewicht befindet, welches sich gemäß
Eine Darstellung der Desorptionsrate als Funktion des Bedeckungsgrades und der Temperatur liefert unter den o. a. Voraussetzungen auf jeden Fall ein genaues und eindeutiges Abbild des Thermodesorptionsvorganges. Zu einer solchen Darstellung gelangt man zwanglos, wenn man bei der numerischen Prozedur der Integration der Desorptionsrate die Wertetripel (Qi; Ti; Ri) bildet. Einem jeden Ratenwert Ri ist somit die momentane Substrattemperatur Ti und -bedeckung Qi zugeordnet.
Die Desorptionsratenfunktion in Abhängigkeit der zwei Variablen Qi und Ti läßt sich dreidimensional darstellen. Dies ist in
Hier sind die TD-Spektren zu verschiedenen Anfangsbedeckungen zu TD-Trajektorien (bzw. -Pfaden) geworden, die den Verlauf der Thermodesorption über die TD-Fläche hinweg beschreiben. Die einzelnen TD-Maxima sind hier sehr schön separiert und können aus verschiedenen Raumrichtungen betrachtet werden. Die Projektionen der Pfade in die Zustandsebene (Q, T) sowie in die Desorptionsratenebenen (Q, R), und (T, R) sind ebenfalls in

| |
| Abbildung A15 Desorptionspfade mit Projektionen in die Zustandsebene und die Ratenebenen Cu/Re(0001) [WaC00/1]. |
|
Die Projektion in die Desorptionsratenebene (T, R) entspricht den ursprünglich gemessenen TD-Spektren. Hieraus ist der Vorteil der dreidimensionalen Darstellung zu erkennen. Innerhalb der normalen TD-Spektren erhält man nur Aussagen über den Verlauf der Desorptionsrate mit der Temperatur, wobei man keinerlei Aussagen über den Bedeckungsgrad machen kann, außer, daß er sich verringert. Der funktionale Zusammenhang in den TD-Spektren läßt sich leichter erkennen, wenn die Desorptionsrate logarithmisch dargestellt wird [ScM92/1, HTS91/1],
|
|
(A70) |
Solange n = 0, Edes und n konstant sind, verlaufen alle Trajektorien eines Zustandes in einer Geraden. Diese nullte Ordnung spiegelt den exponentiellen Verlauf der gemeinsamen Anstiegsflanke der TDS wider. Für diese Abschnitte liefern Steigung und der Achsenabschnitt unmittelbar die Arrheniusparameter Frequenzfaktor ν und Desorptionsenergie Edes (Verfahren nach Polanyi [Wag97/d, Chr91/b]).
Habenschaden und Küppers [Wag97/d, HaK83/1] umgehen die Forderung nach der Konstanz der Parameter, indem sie nur den ersten kleinen Bereich der Desorption untersuchen, in welchem die Änderungen noch nicht ins Gewicht fallen.
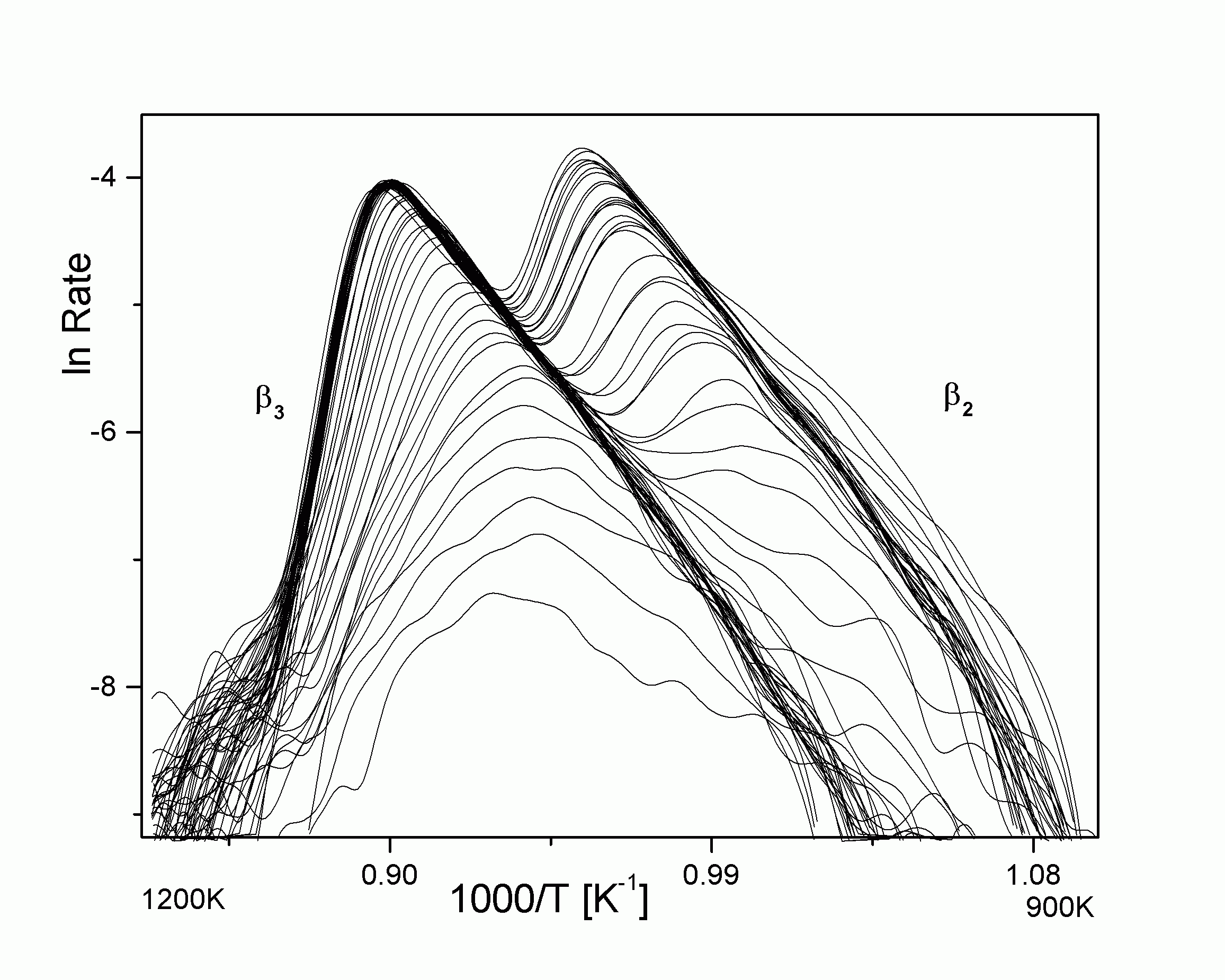
|
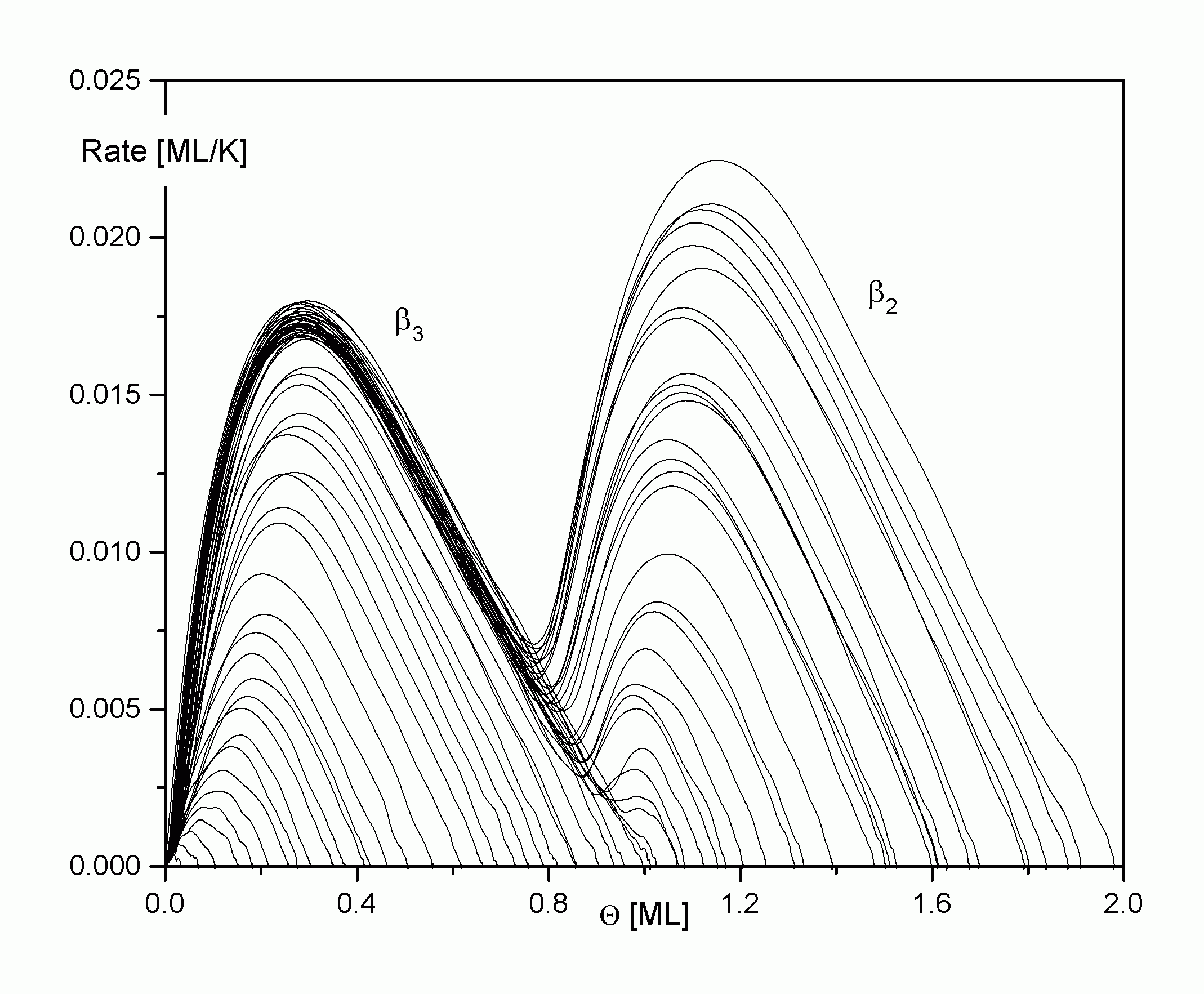
|
||
| Abbildung A16 �lnTDS' Cu/Re(0001) [WaC00/1]. |
|
Abbildung A17 �layer plots' Cu/Re(0001) [WaC00/1]. |
|
Aus der Projektion der Desorptionsfunktion in die andere der beiden Desorptionsratenebenen (Q, R) erhält man die von Menzel et al. eingeführten layer plots [ScM92/1] (
Außer der Desorptionsrate kann auch die zuvor erwähnte Lebenszeit von Adteilchen zur Charakterisierung des Desorptionssystems herangezogen werden [BPT74/1, BaT75/1]. Nach Bauer errechnet sich diese Lebenszeit als Quotient des Bedeckungsgrades und des Massenflusses, in diesem Fall also der Desorptionsrate:
|
|
(A71) |
Nach dem gleichen Prinzip, welches bei der Darstellung der Desorptionsrate als Funktion von Q und T angewendet wurde, ist es möglich, durch Berechnung der Wertetripel (Qi; Ti; ti) die zu den Trajektorien aus
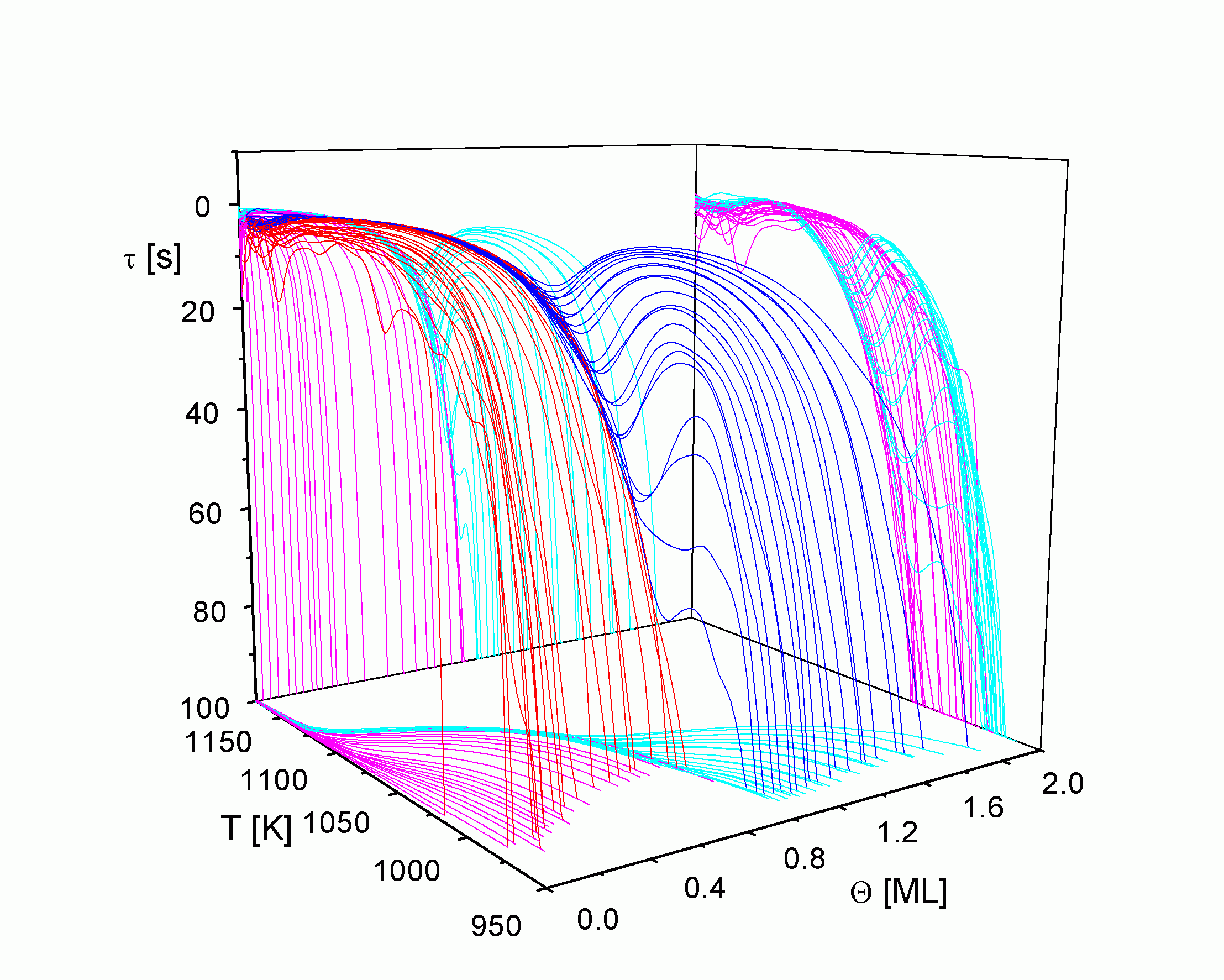
| |
| Abbildung A18 Lebenszeit-Pfade mit Projektionen Cu/Re(0001) [WaC00/1]. |
|
In Kap.
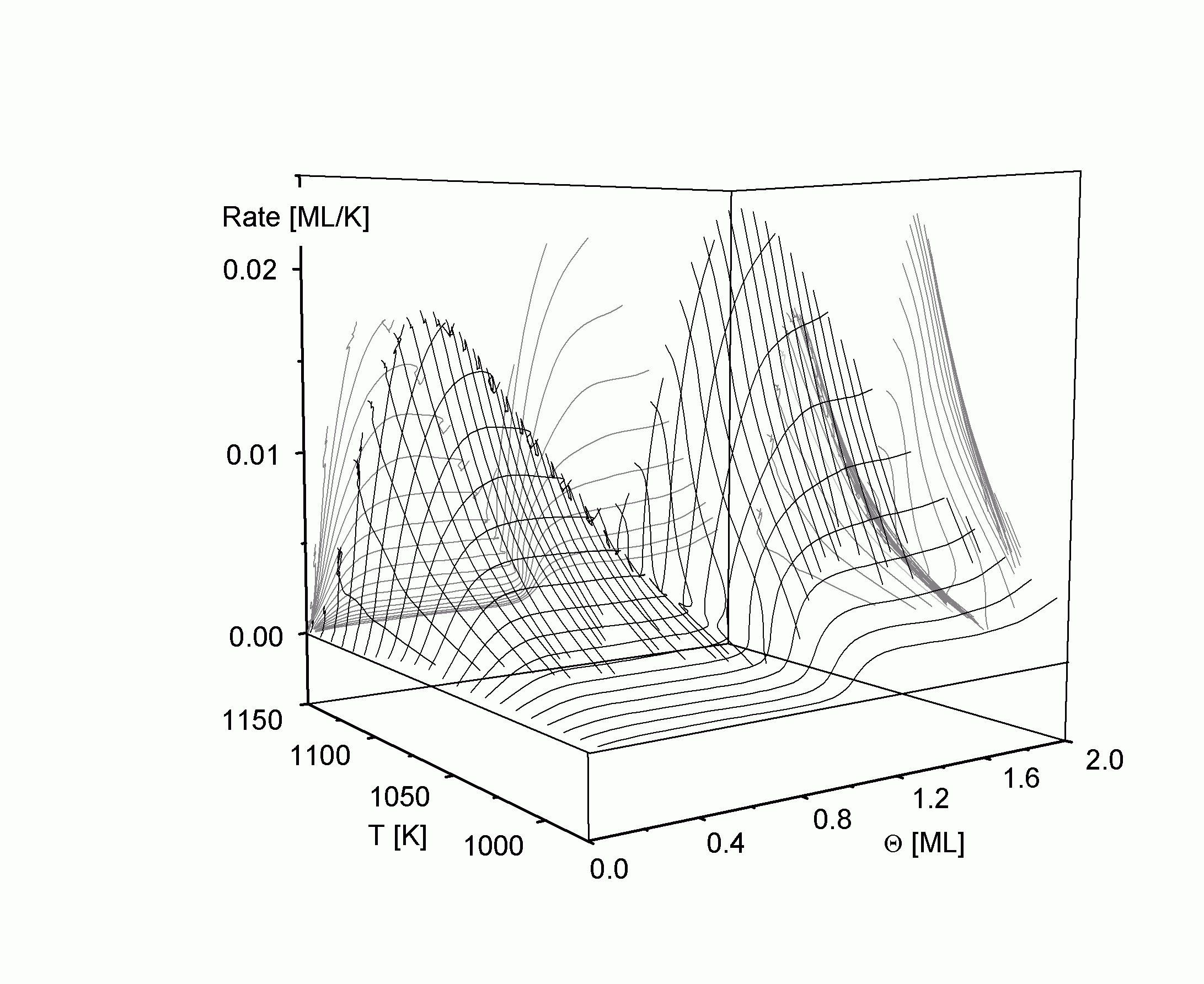
| |
| Abbildung A19 Thermodesorptionsfläche mit Projektionen Cu/Re(0001) nach [WaC00/1]. |
|
Innerhalb der insgesamt gekrümmten TD-Fläche sind Gebiete zu erkennen, die eine in Q-Richtung exponentiell gekrümmte und in T-Richtung ebene Fläche darstellen. Beide Verläufe sind deutliche Hinweise auf eine Desorption nullter Ordnung in diesen Gebieten. Insbesondere dokumentiert sich diese Flächenform in der (Q; R)-Projektion, die die Darstellung der isothermen Desorption mit der Rate Rith ist (
|
|
(A72) |
Bei einer Desorptionsordnung von n = 0 und einem konstanten Funktionsverlauf in diesem Gebiet müssen natürlich auch die Arrheniusparameter ν und Edes konstant sein. Zu vergleichbaren Ergebnissen kommen auch andere Autoren [PFE78/1, PfM83/1, PaB87/1, NaH87/1, Pay88/1, Kre90/1, Kre90/2].
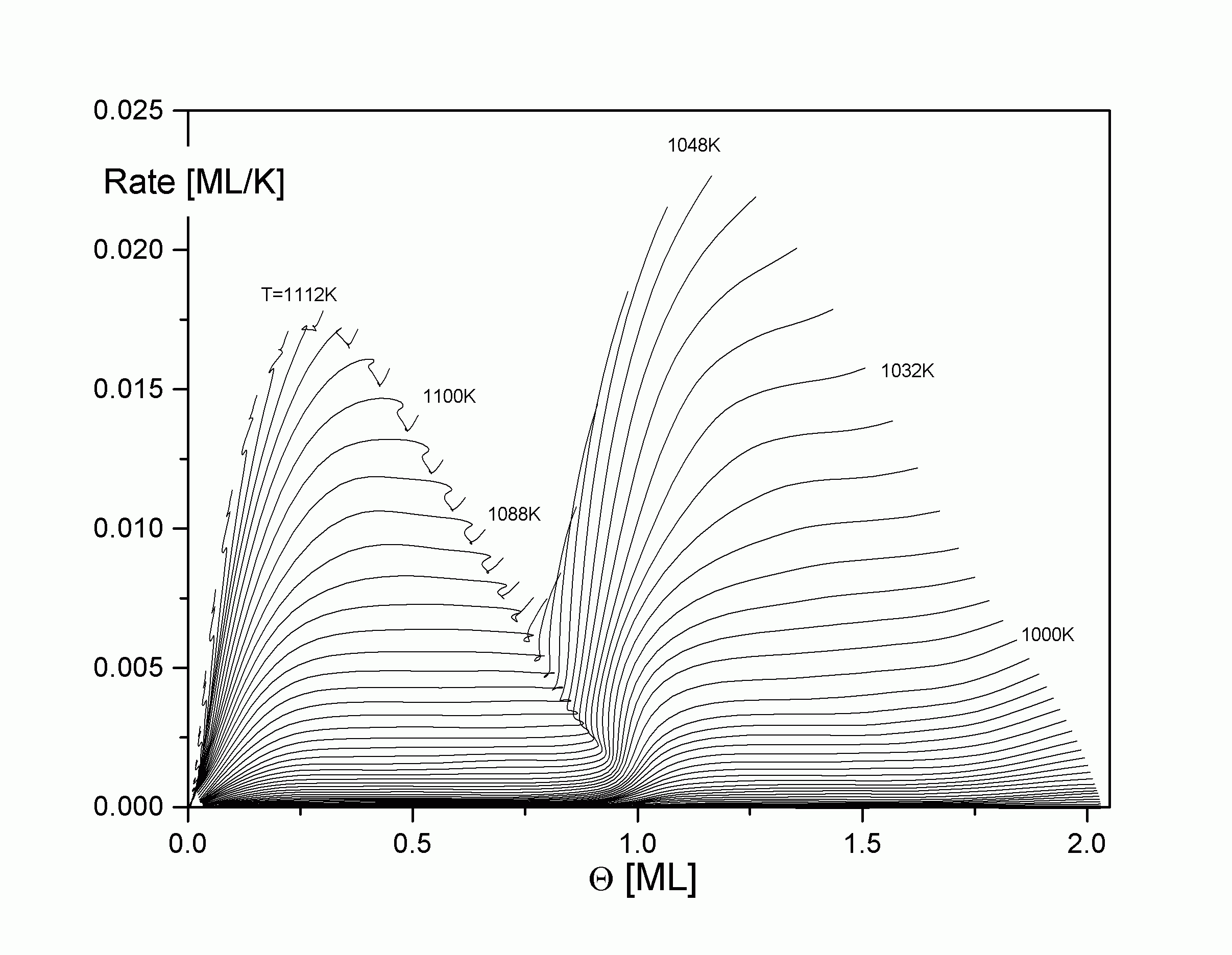
| |
| Abbildung A20 Isothermen Cu/Re(0001) [WaC00/1] |
|
Bei der Formulierung einer isothermen Desorptionsrate (also einer Desorption bei konstanter Temperatur) erhebt sich die Frage, wie ein derartiges Experiment praktisch angestellt werden kann. Tatsächlich wurden solche Experimente durchgeführt. Bauer et al [PaB87/1, PaB87/2, PSB88/1] haben die direkt gemessenen isothermen Ratenwerte mit denen aus TD- Spektren erhaltenen verglichen und eine gute Übereinstimmung festgestellt. Dabei wurde die Probe bei einer konstanten Temperatur, bei der bereits Desorption stattfindet, gehalten und mit Adsorbatmaterial bedampft. Zu einem bestimmten Zeitpunkt wurde der Teilchenstrom abgeschaltet und die Abnahme des Bedeckungsgrades bei dieser Temperatur verfolgt, die sich in der Veränderung des Teilchenstroms manifestiert.
Eine interessante Möglichkeit, weitere Eigenschaften des Desorptionssystems zu bestimmen, ergibt sich, wenn man die Isothermen
|
|
(A73) |
In
In der Arbeit von Wagner [WSC99/1] sind order plots des Systems Cu/Re(0001) bis zu einem Bedeckungsgrad von ca. 6 ML dargestellt. Es sind dort hauptsächlich vier Abschnitte zu erkennen, die einer gemeinsamen Steigung zuzuordnen sind. Besonders klar sind die Verhältnisse bei den Isothermen geringerer Temperatur. Diese haben drei Abschnitte, in denen der Anstieg Null ist. Im letzten Teil (Q < 0,15 ML) ist der Anstieg etwa eins. Die anderen Isothermen zeigen das gleiche Verhalten, mit der Einschränkung, daß mit steigender Temperatur die Anzahl der Gebiete mit konstanter Steigung abnimmt. Dies ist verständlich, wenn man bedenkt, daß mit ansteigender Temperatur mehr und mehr Zustände desorbieren und zur Darstellung nicht mehr beitragen können.
Es ist also festzustellen, daß beim hier beispielhaft gewählten System Cu/Re(0001) für den Bereich mit Q < 0,15 ML eine Desorption erster Ordnung vorliegt. Ab Q = 0,15 ML wird die Desorptionsordnung Null und bleibt dies so lange, bis eine Bedeckung von Q = 1 ML erreicht ist. Diese Tatsache kann mit einem bereits in
Der Verlauf für die dritte bis n-te Lage (Multilage) ist ebenfalls parallel zur Q-Achse. Vom horizontalen Teil der Kurve für die zweite Lage ist er durch einen Sprung, äquivalent dem vom Übergang von der ersten zur zweiten Lage getrennt. Bei weiteren Lagenübergängen (3®4, 4®5, usw.) treten allerdings keine Sprünge mehr auf.
Aus dem Verlauf der order plots bei n = 0 ist ein Kompensationseffekt zwischen Präexponential- und Exponentialterm abzuleiten. Setzt man nämlich lnRith = const. und n = 0 in Gleichung (A73) ein, erhält man:
|
|
(A74) |
Diese Desorption nach nullter Ordnung ist somit ein Zeichen für den Kompensationseffekt, also den gleichen Verlauf von lnn und Edes mit dem Bedeckungsgrad.
Eine weitere Projektionsmöglichkeit der TD-Funktion ist die in die Desorptionsratenebene (T, R). Dadurch erhält man die in
Auch bzw. gerade hier stellt sich die Frage nach einer experimentellen Realisierung:. Die Desorptionsrate ist ja gerade als Änderung des Bedeckungsgrades definiert. Diese Auffassung müßte man für eine experimentelle Durchführung insoweit ändern, als daß die Desorptionsrate jetzt mit dem sich von der Probe weg bewegendem Partikelstrom identifiziert wird. Praktisch müßte dieser Partikelstrom detektiert werden und gleichzeitig durch einen entsprechenden entgegengerichteten Partikelstrom zum Konstanthalten des Bedeckungsgrades kompensiert werden. Dies könnte durch ein geregeltes Bedampfen geschehen. Dabei müßte wie beim eigentlichen TD-Experiment die Temperatur linear erhöht werden.
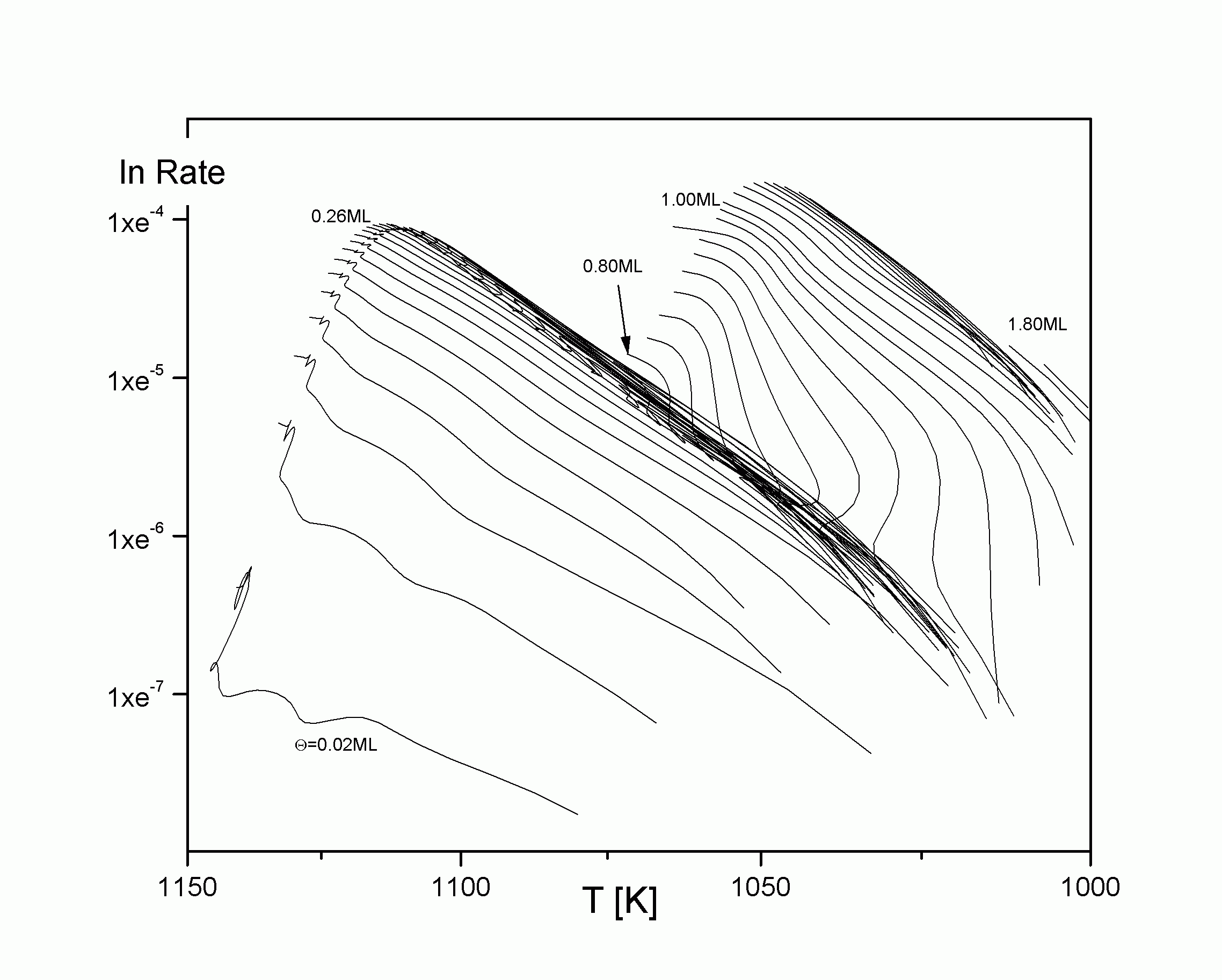
| |
| Abbildung A21 Arrhenius-plots Cu/Re(0001) [WaC00/1] |
|
Auch die Darstellung der Isosteren hat eine weitere wichtige Anwendung. Stellt man nämlich die Bedeckungsgradachse logarithmisch und die Temperaturachse reziprok dar, erhält man die Arrhenius-plots von
|
|
(A75) |
Bauer et al. [BPT74/1, BPT75/1, KoB86/2] haben ein Verfahren zur Ermittlung der Desorptionsenergie und des dazugehörigen Frequenzfaktors entwickelt, bei dem sie sich des Arrheniusansatzes (A69) bedienen. Allerdings benutzen sie nicht Desorptionsratenisosteren sondern Lebenszeitisosteren. Diese werden auf graphischem Wege aus Lebenszeitspektren (vgl.
|
|
(A76) |
Ein numerisches Verfahren nach Schlatterbeck [Sch98/d], das sich an die Bauersche Methode anlehnt, wird in [WSC99/1] verwendet, um die Bedeckungsgradabhängigkeit der Desorptionsenergie mit hoher Genauigkeit zu ermitteln. Die Ergebnisse dieses Verfahrens unterscheiden sich nicht von denen, die nach King [Kin75/1] durch Nutzung der Desorptionsrate anstelle der Lebenszeit erhalten werden.
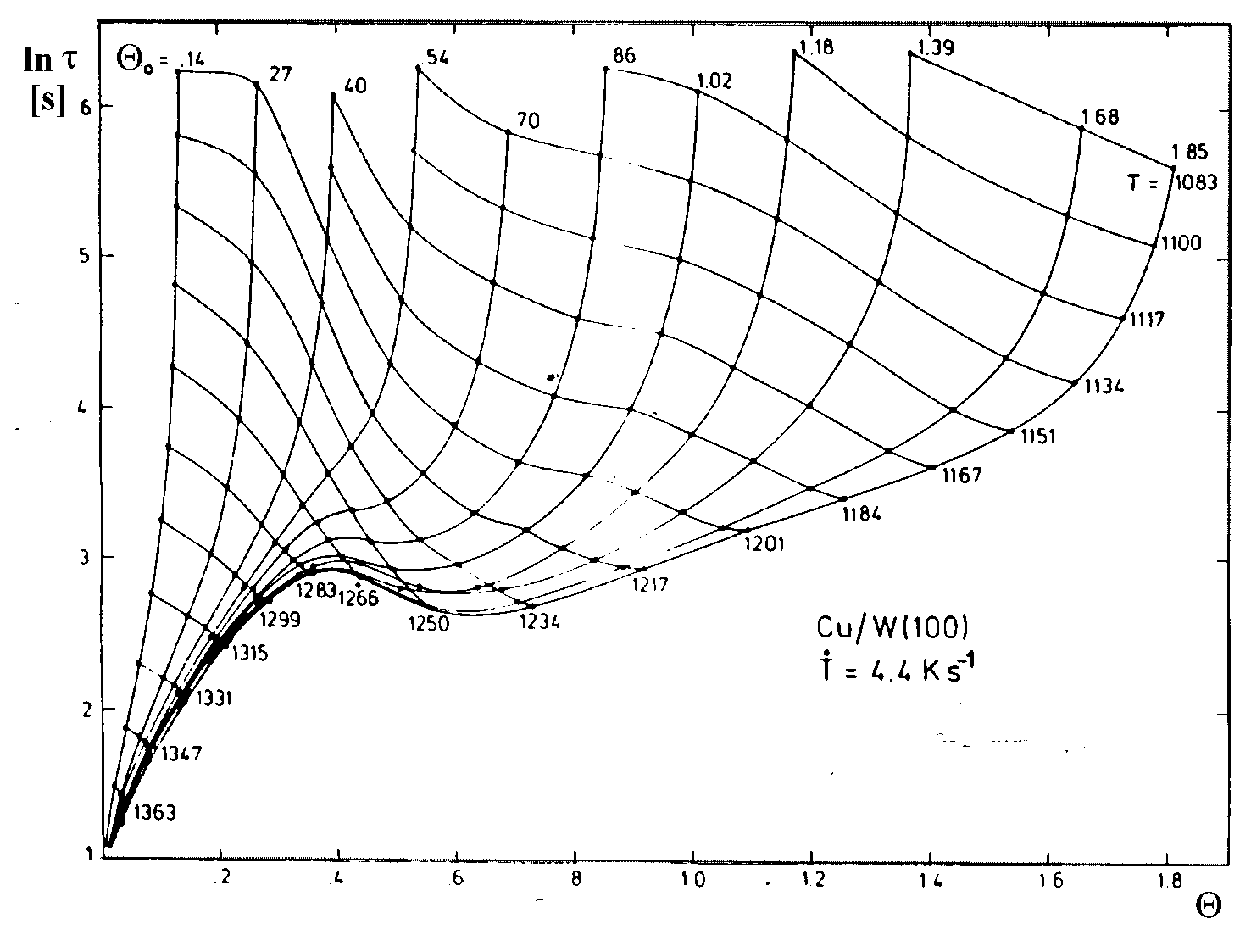
| |
| Abbildung A22 Lebensdauer und dazugehörige Isosteren von Cu/W(110) [BPT75/1] |
|
Wenn dem eigentlichen Desorptionsprozeß komplizierte physikalische Vorgänge überlagert sind, weisen die Arrhenius-plots gekrümmte Abschnitte oder auch Knicke auf.
Dies kann mehrere Ursachen haben [BPT75/1, Wag97/d], wie z. B.
Besonders im Submonolagenbereich erhält man z.T. die beste Übereinstimmung durch das Zuordnen von zwei Arrheniusgeraden, eine im Bereich sehr kleiner Bedeckungen für den Zustand des 2D-Gases und eine für die 2D-Gas-Kondensat-Koexistenzregion bei höheren Bedeckungen [BaP87/1].
Als weitere Auswertungsmethoden zur Ermittelung der Desorptionsenergie aus TD-Spektren, kommen folgende in Betracht (Einzelheiten können der angegebenen Literatur entnommen werden.):
Die meisten Auswertungsmethoden für TDS gründen sich auf die Polanyi-Wigner-Gleichung und extrahieren die "Desorptionsenergie" und den "Frequenzfaktor der Desorption" z. T. als konstante Funktionen oder als Funktionen des Bedeckungsgrades aus einzelnen TD-Spektren oder TD-Spektrenserien. Dies mag für sehr einfache Systeme möglich und richtig sein. Treten jedoch im durch die TDS spektroskopierten Bereich (T, Q ) andere Prozesse auf, deren Raten nicht durch die Polanyi-Wigner-Gleichung beschrieben werden, müssen weitergehende Analysen vorgenommen werden. So kann der Desorptionsprozeß z. B. durch ein vorgelagertes 2D-Phasengleichgewicht beeinflußt sein.
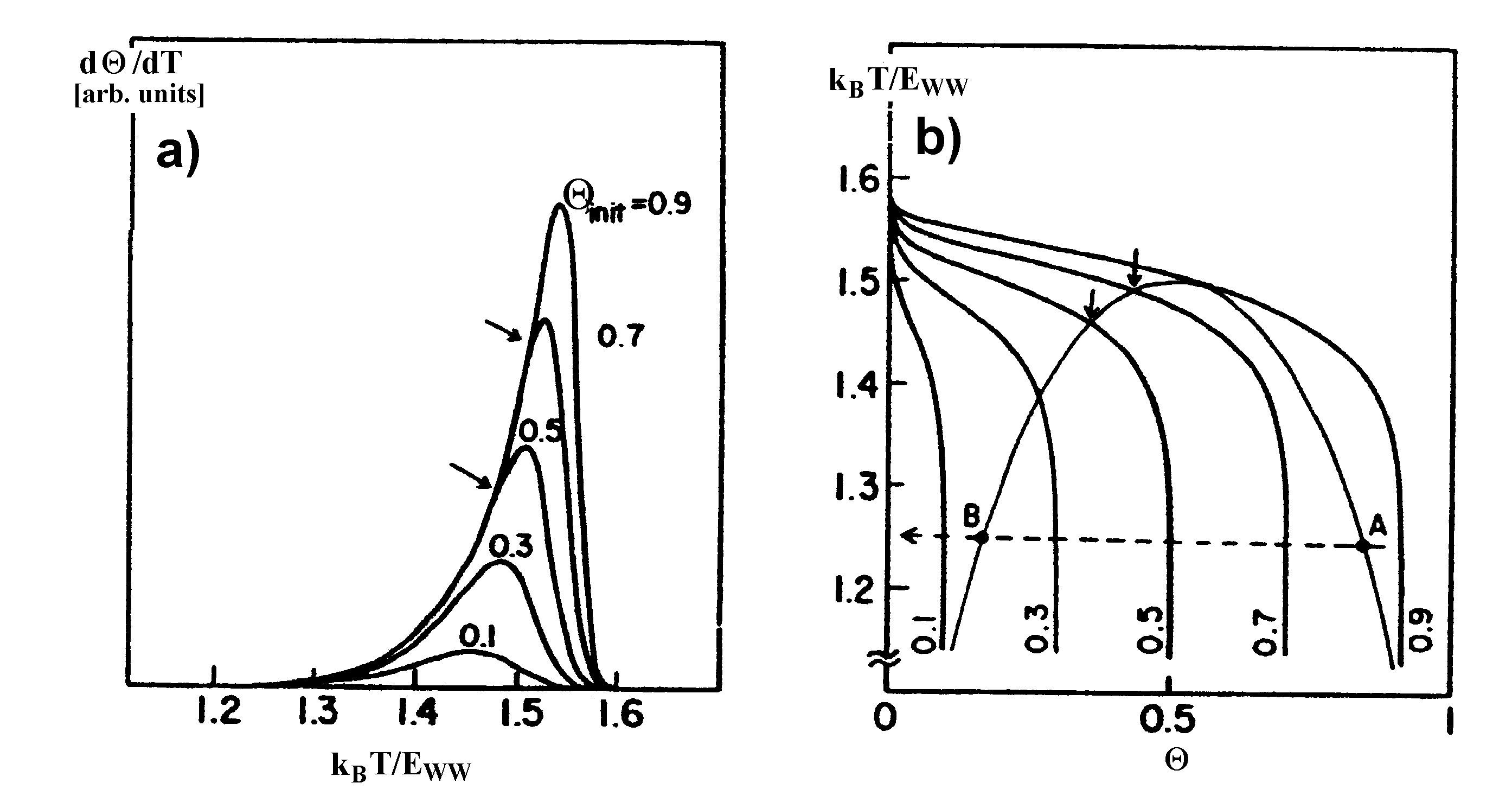
| |
| Abbildung A23 a) TDS und b) T=�(Q) als Desorptionspfade im Phasendiagramm [Nag86/1]. |
|
Nagai [Nag85/1, Nag85/2, Nag86/1] stellt einen direkten Bezug zwischen der Form von TD-Spektren und einem solchen 2D-Phasenübergang (2D-Inseln � 2D-Gas) her. Er formuliert unter Bezug auf die Bragg-Williams-Näherung (vgl.
| |
| Abbildung A24 Desorptionsisothermen und Lage der Phasengrenze nach [EGC86/1]. |
|
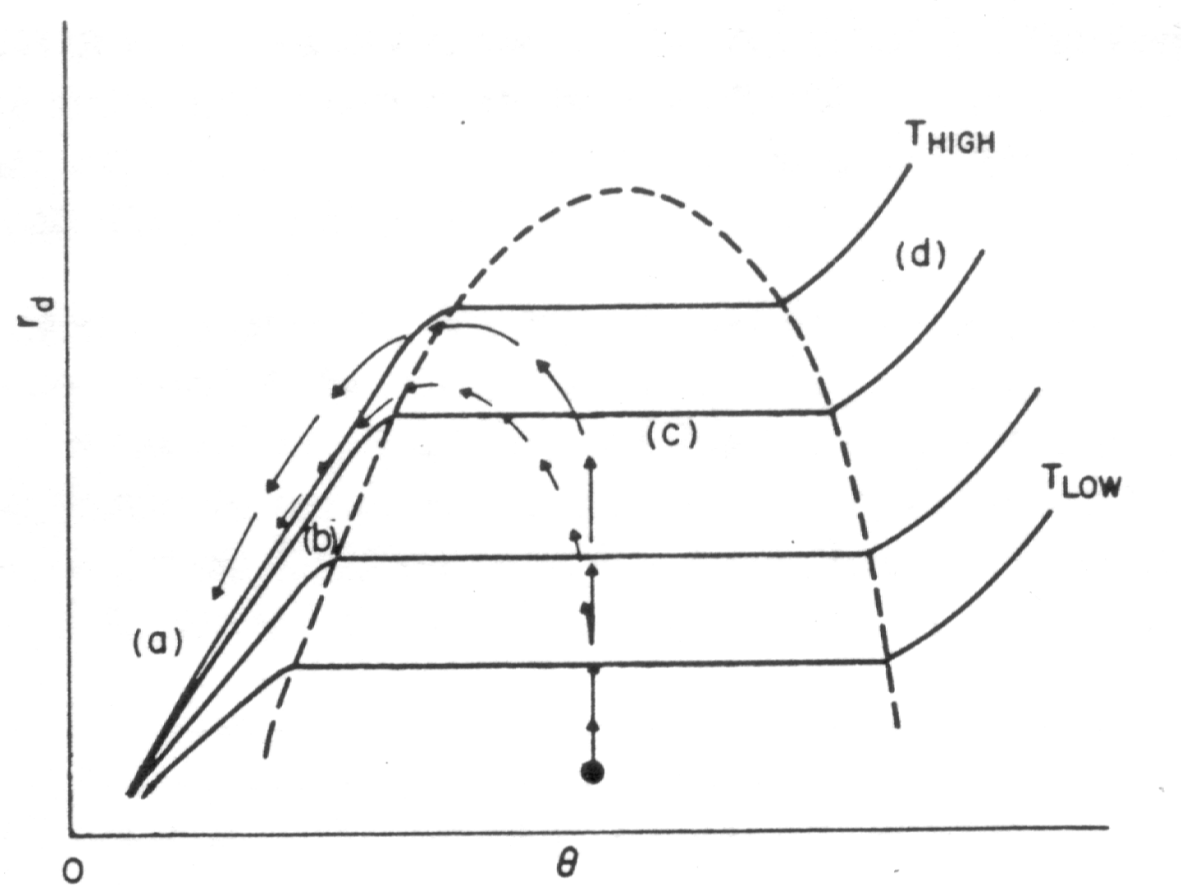
Eingezeichnet ins Phasendiagramm sind die Desorptionspfade der Thermodesorptionsspektren. Die Pfeile markieren den Übergang von der Desorption nullter zur ersten Ordnung. Im Desorptionsspektrum sind gerade das die Punkte, an denen der gemeinsame Anstieg endet und die Spektren auseinanderlaufen.
Estrup et al. zeigt, wie anhand des Verlaufes von Desorptionsisothermen (vgl.
Die Berücksichtigung der Bragg-Williams-Näherung (vgl.
Kreuzer et al. betrachten unter Nutzung eines speziellen Mehrlagen-Gittergasmodells und mit Hilfe verschiedener Näherungen, wie BWA, QCA u. a., diverse Adsorptions- und Desorptionskinetiken, die sie mittels Transfermatrixmethoden berechnen [PaK95/1]. So können sie sowohl Desorptionsrate als auch Desorptionsenergie und andere Meßgrößen simulieren und erhalten viele systembestimmenden physikalischen Größen, wie EWW, Vz,0, vgl.
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}