|
|
|
Kapitel A2 | Abschnitt A - Theoretische Grundlagen |
|
|
|
Kapitel A4 | Kapitel A3 - Kinetik und Energetik von Oberflächenprozessen |
3. Kinetik und Energetik von Oberflächenprozessen
Bisher wurden die Prozesse des Auftreffens des Metallatoms aus der Gasphase auf die Substratoberfläche sowie des Wachstums epitaktischer binärer Metallsysteme betrachtet. Doch wie schon in
Als Prozesse auf der Oberfläche, die zu einer Änderung der Bedeckung führen, kommen die Desorption (mit der Desorptionsrate
|
|
(A13) |
){kind=link}
Eine untergeordnete Rolle spielen Diffusionsprozesse zum Rand oder zur Rückseite sowie ins Volumen des Substrates. Die Stabilität eines Zustandes kann man über die "Lebensdauer" des dazugehörigen Teilchens auf der Oberfläche definieren [BaT75/1]:
|
|
(A14) |
){kind=link}
Bezüglich der o. a. Raten können drei Fälle grob unterschieden werden:
3.1. Kinetik der Desorption
Zur Desorption muß den Adteilchen, welche desorbieren sollen, genügend Energie zugeführt werden. Dies kann in Form von Wärme geschehen, also durch Erhitzen der Probe, aber auch beispielsweise beim Einstrahlen von Laserlicht oder durch Elektronenstoß. Weiterhin benötigt man eine Meßmethode, um die Vorgänge zu verfolgen. Es bietet sich hier die Thermodesorptionsspektroskopie (TDS) an, bei der die Prozesse der thermischen Anregung und der Untersuchung miteinander verknüpft sind.
Wendet man also die TDS zur Untersuchung von Adsorbatsystemen an, werden diese in dem erhöhten Temperaturbereich vom Einsetzen bis zum Abklingen der Desorption betrachtet. Die Desorption wird durch thermische Energie RT, die dem System über eine lineare Heizrampe (
Die verschiedenen möglichen Adsorptionsplätze wurden bereits in
Mit hoher Wahrscheinlichkeit werden nur bestimmte, ausgezeichnete Adsorptionsplätze als Desorptionsplätze fungieren. Im Rahmen einfacher energetischer Vorstellungen sollten die am schwächsten gebundenen Teilchen zuerst und die am stärksten gebundenen zuletzt desorbieren. Insofern sind Adteilchen, die sich frei auf der Oberfläche bewegen bzw. schwach bindende Adsorptionsplätze bevölkern, potentiell die ersten Teilchen, die desorbieren werden. Andererseits entsprechen hochkoordinierte Adsorptionsplätze wie Defekte (insbesondere Stufen, Fehlstellen) Zentren mit besonderer Bindungsfähigkeit, von denen aus die Teilchen zuletzt desorbieren werden. Adinsel- und Adinselrandplätze sind ebenfalls hochkoordiniert, diese Plätze können allerdings in der Endphase der Desorption, wenn es keine Inseln mehr gibt, überhaupt nicht existent sein.
Viele Theorien zur Beschreibung der Desorptionsprozesse berücksichtigen nicht die Wechselwirkung der Adteilchen untereinander und behandeln das System, als ob es sich um ein auf der Oberfläche befindliches ideales Gas handeln würde. In der Realität bestehen aber, zumindest ab einem bestimmten Bedeckungsgrad, sehr wohl Wechselwirkungen zwischen den Adteilchen, und die o. a. Überlegungen gelten deshalb nur im Bereich sehr kleiner Bedeckungsgrade [SKS88/1]. Andere Theorien gehen von einem Quasigleichgewichtszustand während der Desorption aus und vernachlässigen zunehmend die kinetische Seite des Desorptionsprozesses. Allerdings ist man hier in der Lage, viele zusätzlich zur Desorption auftretende Phänomene zu beschreiben. In den nächsten zwei Kapiteln sollen Beispiele der kinetischen und der energetische Herangehensweise näher beschrieben werden.
3.2. Theorie des Übergangszustandes
Eine einfache Theorie zur Beschreibung der Desorptionsvorgänge geht auf die Erkenntnisse von Eyring [GLE41/b] zurück, der kinetische Prozesse mittels der Theorie des Übergangszustandes (Transition State Theory, TST) als einen Vorgang beschrieb, bei dem während der Reaktion in einem vorgelagerten Gleichgewicht ein aktivierter Komplex gebildet wird. Dieser Komplex reagiert dann in einem irreversiblen Prozeß zum Reaktionsprodukt, dessen Rate durch die Zustandsdichte am Übergangszustand bestimmt wird. Der Übergangszustand befindet sich dabei am Potentialmaximum oder in seiner Nähe. (Die Energieniveaus sind dabei thermisch, d.h. nach einer Boltzmann-Verteilung besetzt.) Wendet man die TST auf den Prozeß der Thermodesorption an, so spricht man in diesem Fall von der Theorie der Absoluten Rate (Absolute Rate Theory, ART).
Das vorgelagerte Gleichgewicht kann man mit Hilfe des Massenwirkungsgesetzes (MWG) wie folgt formulieren (Z ... Zustandssumme, D E0 ... Energiedifferenz zwischen Grundzustand) [Chr91/b]:
|
|
(A15) |
){kind=link}
Nach der Umwandlung eines Freiheitsgrades der Schwingung des "aktivierten Komplexes" (gegen die Oberfläche) in einen Translationsfreiheitsgrad der Reaktion (Separation der Nullpunktsenergie) erhält man für die Gleichgewichtskonstante:
|
|
(A16) |
){kind=link}
und für die Reaktionsrate mit
|
|
(A17) |
){kind=link}
|
|
(A18) |
){kind=link}
k ... Reaktionsgeschwindigkeitskonstante, Z ... Zustandssumme, Edes. ... Desorptionsenergie. (Der Transmissionskoeffizient k berücksichtigt quantenmechanische Prozesse wie z. B. das Untertunneln der Aktivierungsbarriere oder auch die Rückreaktion des Übergangszustandes ‡. Bei der Metall-Desorption ist k = 1 eine gute Näherung.)
Ganz ähnlich können die Vorgänge bei der Desorption von Atomen aus Metallschichten von Metall-Einkristalloberflächen beschrieben werden. Die Bildung des aktivierten Komplexes ist im Rahmen der Theorie des Übergangszustandes etwas subtil; es wird hier auf die Erkenntnisse aus anderen Näherungen, die in
Die Desorptionsrate ergibt sich nach
|
|
(A19) |
){kind=link}
mit Q ... Bedeckungsgrad, n ... Desorptionsordnung zu
|
|
(A20) |
){kind=link}
Hierbei gilt:
|
|
(A21) |
){kind=link}
und
|
|
(A22) |
){kind=link}
ndes. ... Frequenzfaktor der Desorption, D Sdes ... Desorptionsentropie [BPT75/1, SKS88/1, Zhd91/1].
Ein Teilchen sollte um so schneller von der Oberfläche desorbieren, je größer der Entropiegewinn dabei ist. Dieser ist für die Desorption aus einer dichteren Phase (also mit höherem Vorfaktor, s.u.) größer als der für die Desorption aus einer weniger dichten Phase. Für fest gebundene Teilchen ist demnach der entropische Beitrag zur Triebkraft der Desorption größer als für nur schwach gebundene.
Setzt man νdes aus (A21) in (A20) ein erhält man so die Polanyi-Wigner-Gleichung,
|
|
(A23) |
){kind=link}
und
|
|
(A24) |
){kind=link}
Bei energetisch unterschiedlichen Adsorptionsplätzen sind mehrere derartige Ratengleichungen nötig. Wenn die Teilchen zwischen Adsorptionsplätzen wechseln können, kann es nötig werden, zusätzlich einen Diffusionsterm zu berücksichtigen.
Die Zustandssumme in (A21) setzt sich allgemein aus den Anteilen der Translation, der Rotation und der Vibration auf bzw. gegen die Substratoberfläche sowie den entsprechenden internen Bestandteilen zusammen.
|
|
(A25) |
){kind=link}
Atome besitzen keinen Rotations- und keinen internen Freiheitsgrad. Es gilt also
Die Teilchen des Grundzustandes sollen durch das Bindungspotential des Systems lokal fixiert sein, und es gilt also Zt = 1. Die Teilchen des Übergangszustandes hingegen besitzen genügend Energie, um sich frei auf der Oberfläche bewegen zu können:
|
|
(A26) |
){kind=link}
Man kann den Teilchen auch eine thermische Wellenlänge zuordnen, die wie folgt definiert ist:
|
|
(A27) |
){kind=link}
also
|
|
(A28) |
){kind=link}
m ... Masse des Adteilchens, N ... Anzahl der Adteilchen auf der Substratfläche, l... thermische Wellenlänge der Adteilchen [Zhd81/1].
Die Desorptionsrate hat jetzt die Form
|
|
(A29) |
){kind=link}
Durch Verwendung der Hochtemperaturnäherung kBT >> hn ergibt sich
|
|
(A30) |
){kind=link}
und mit den Annahmen, daß erstens das Bindungspotential Vz und damit auch nz für den Übergangs- und Grundzustand gleich sind und daß zweitens auf Grund der freien Beweglichkeit der Teilchen im Übergangszustand
|
|
(A31) |
){kind=link}
für ein System, bei dem die Teilchen des Grundzustandes durch das Oberflächenpotential lokalisiert sind. Sind diese Teilchen jedoch ebenfalls frei auf der Oberfläche beweglich, wird Z‡ = Z und damit
|
|
(A32) |
){kind=link}
|
|
(A33) |
){kind=link}
Aus der Annahme, daß t bedeckungsgradunabhängig ist, leitet Bauer [BaT75/1] ab, daß n und Edes zwar nicht auch bedeckungsgradunabhängig sind, daß aber wohl zwischen Edes und lnn ein linearer Zusammenhang, d.h. ein Kompensationseffekt besteht. Wandelt erklärt, daß (für die Fälle, in denen ein Kompensationseffekt besteht) dieser daraus resultiert, daß mit steigender Desorptionsenergie, die die Tiefe des Bindungspotentials darstellt, auch die Krümmung dieser Kurve (die durch den Vorfaktor repräsentiert wird) zunimmt [NMW88/1]. Kreuzer erklärt den Kompensationseffekt während 2D-Phasenübergängen mit der Konstanz des chemischen Potentials und der damit verbundenen Kompensation enthalpischer und entropischer Beiträge (vgl. Gl. (A50), (A51) im Zusammenhang mit (A20), (A21) und (A22)) [KrM88/1]. Eine gründliche Betrachtung der Vorgänge im Zusammenhang mit dem Kompensationseffekt ist bei Seebauer et al. zu finden [SKS88/1].
){kind=link}
){kind=link}
3.3. Das Modell des 2D-Gases
Adsorptionssysteme mit einer kristallographisch (und energetisch) glatten Oberfläche (wie die Re(0001)-Oberfläche) können besonders gut mit Hilfe des Modells des 2D-Gases beschrieben werden. Dieses Modell basiert auf zwei Adsorbat-Anteilen, die sich im thermodynamischen Gleichgewicht befinden.
Der eine Anteil ist in Adsorbatinseln lokalisiert, der andere kann sich auf der blanken Substratoberfläche frei (vom Potential in x- und y-Richtung) bewegen und wird als 2D-Gas angesehen, das ideale Eigenschaften besitzen soll. (Tatsächlich besitzen die Adteilchen nur für Q ® 0 ideale (2D-) Gaseigenschaften. Bei endlichen Bedeckungsgraden treten vermehrt interne Wechselwirkungen zwischen den Adteilchen auf, und das ideale wird zum realen System. Zusätzliche zu einer völlig freien Oberflächendiffusion kann es zu einer Art Hüpfvorgang kommen. Dabei verbringen die Adteilchen eine gewisse Zeit auf einem Adsorptionsplatz und springen dann auf einen Nachbarplatz. Die Bewegung der Teilchen verursacht eine insgesamt schwächere Wechselwirkung mit dem Substrat, da sich die Teilchen im zeitlichen Mittel auch eine gewisse Zeit auf ungünstigen Adsorptionsplätzen befinden.)
Der kondensierte Anteil der Adteilchen kann hingegen z. B. mit dem Einstein-Modell beschrieben werden. (Dieses Modell beschreibt den Festkörper als ein System harmonischer Oszillatoren bestehen, deren Schwingungsfrequenz durch n gegeben ist.)
Die hier betrachtete Re(0001)-Substratoberfläche mit ihrer genau definierte Anzahl von Adsorptionsplätzen Nmax ist mit einer bestimmten Anzahl (
Das Modell des 2D-Gases beschränkt sich auf die Vorgänge innerhalb einer Lage. Damit kann man die Teilchendichte durch den Bedeckungsgrad dieser Monolage Q = NA/N ausdrücken. Q kann maximal 1 ML sein, wenn jeder Adsorptionsplatz durch genau ein Teilchen besetzt ist.
Bei geringen Bedeckungen oder hohen Temperaturen sind alle Teilchen auf der Oberfläche delokalisiert und bilden eine reine 2D-Gasphase (oder auch 2D-Flüssigkeit) aus. Bei einem bestimmten (Grenz-) Bedeckungsgrad, den wir mit QG bezeichnen wollen und der von der Temperatur abhängt, werden die attraktiven Wechselwirkungen wegen der zunehmenden räumlichen Nähe der Teilchen sehr stark. Damit nimmt auch die Wahrscheinlichkeit zur Bildung von stabilen Kondensationskeimen zu. Dies hat zur Folge, daß Teilchen kondensieren und eine neue Phase bilden. Diese besteht aus Inseln auf der Oberfläche, zwischen denen sich weiterhin 2D-Gasteilchen bewegen können. Unterhalb der kritischen Temperatur Tc stehen im Zweiphasengebiet die Kondensat- und 2D-Gasteilchen, wie o. a., im Gleichgewicht [Per92/1].
Bei gleichbleibender Temperatur entspricht die 2D-Gaskonzentration derjenigen bei QG. Die Inseln bilden eine einheitliche thermodynamische Phase. Die Kondensatkonzentration ist somit (wie etwa im dreidimensionalen Fall der Fällung eines schwerlöslichen Stoffes oder dem Erstarren einer Flüssigkeit) eins. Die Bedeckung mit kondensierten Teilchen Qc hängt jedoch von der Anzahl und der Größe der Inseln ab. Durch einen steigenden Anteil von Kondensatteilchen (etwa bei Adsorption) wird der dem 2D-Gas zur Verfügung stehende Raum kleiner, weshalb sich auch bei konstanter "Gasdichte" der Flächenanteil der 2D-Gas-Spezies verringert.
Grundsätzlich ist die Desorption sowohl aus der 2D-Gasphase mit der Teilrate rg (2D-Gas) als auch aus der kondensierten Phase mit der Teilrate rc (2D-condensed) möglich (R = rg + rc). Die Desorption aus der 2D-Gasphase läuft auf Grund der geringeren Bindung der 2D-Gasteilchen mit einer viel höheren Geschwindigkeit als die der Kondensatteilchen ab (rg >> rc). Somit kann die Desorption direkt aus der kondensierten Phase beim Vorhandensein der 2D-Gasphase vernachlässigt werden und es gälte R ≡ rg.
Weiterhin wird die zweidimensionale Teilrate rcg definiert, die den Übergang von Teilchen der Kondensatphase in die 2D-Gasphase beschreibt. Mit diesen Voraussetzungen können zwei Fälle unterschieden werden [Kre91/b, PaK88/1, KrP88/1]:
rcg << R: Der Übergang vom Kondensat ins 2D-Gas (rcg) ist der langsamste und damit geschwindigkeitsbestimmende Schritt. Die Desorption vollzieht sich hier quasi von den Inselrändern her. Daraus folgt eine "effektive" gebrochenzahlige Desorptionsordnung [Kre91/b, PaK88/1]. Hier kann nicht von einem Gleichgewichtszustand ausgegangen werden. Dieser Fall soll hier nicht weiter vertieft werden.
rcg >> R: Der Übergang ins 2D-Gas ist so schnell, daß ein "Quasigleichgewicht" zwischen den beiden Phasen besteht. Solange kondensierte Teilchen vorhanden sind, werden durch das o. a. vorgelagerte Gleichgewicht 2D-Gasteilchen sofort nachgebildet, wodurch die 2D-Gaskonzentration konstant bleibt. (Diffusionsprozesse sind wegen ihrer hohen Geschwindigkeit nicht ratenbestimmend.) Dieser Fall wird angenommen.
(Bauer hingegen nimmt auch für den Submonolagenbereich eine Desorption aus der kondensierten Phase an, die aus Teilchen der 2D-Gasphase nachgebildet werden[BaT75/1]. Auch hier wird das Gleichgewicht vorausgesetzt, und eine Desorption nullter Ordnung resultiert.)
Die Desorption soll nach unserem Modell nur aus der 2D-Gasphase mit der Rate R möglich sein. Im Bereich sehr kleiner Bedeckungen sind nur 2D-Gasteilchen vorhanden, die direkt desorbieren können. Sind sowohl Gas- als auch Inselteilchen vorhanden, gehen die letztgenannten zunächst mit rcg zuerst in die 2D-Gasphase über, und desorbieren von dort in die 3D-Gasphase [Kre91/b, PaK88/1].
Wir können die Vorgänge bei der Temperaturerhöhung eines solchen Adsorbatsystems (rcg >> R) also als eine Reaktion mit vorgelagertem Gleichgewicht auffassen:
(C = Teilchen der kondensierten Phase, G = Teilchen der 2D-Gasphase und 3D = Teilchen der 3D-Gasphase.)
3.4. Thermodynamik der Desorption
Bei der Untersuchung von Adsorbatsystemen mit internen Wechselwirkungen, wie insbesondere der Thermodesorptionsspektroskopie von Metall-auf-Metall-Systemen, stellt sich heraus, daß diese nicht durch so einfache Zusammenhänge wie beispielsweise die Polanyi-Wigner-Gleichung (A23) beschreibbar sind. Es treten nämlich mehr systembestimmende physikalische Größen auf, die zudem vom Bedeckungsgrad oder von der Temperatur abhängig sind. Um diese zu erhalten und das Meßsystem richtig zu beschreiben, erscheint es sinnvoll, aus den verschiedenen Ansätzen zur Beschreibung des energetischen (thermodynamischen) Zustandes einer adsorbierten Phase denjenigen auszuwählen, der einerseits das System gut beschreibt und andererseits möglichst einfach zu berechnen ist.
Da alle Oberflächenphänomene durch einen solchen Ansatz nicht erfaßt werden können, sollen in den weiteren Betrachtungen einige Voraussetzungen gegeben sein:
- Substrat- und Adatome sind gleich groß, d. h. es besteht kein (Gitter-) misfit [Saf94/b]. (Dieser muß später extra berechnet werden.)
- Das System befindet sich im Quasigleichgewicht. (Diffusionsprozesse sind so schnell, daß sie keinen Einfluß auf die Teilchenverteilung nehmen, s.
Kap. 3.3 .) - Der Haftkoeffizient für Metall-auf-Metall-Systeme ist S = 1, d. h., jedes auf der Oberfläche auftreffende Atom bleibt haften [Kre91/b].
- Bei Metallatomen handelt sich um punktförmige Teilchen ohne innere Struktur.
Eine der einfachsten Möglichkeiten, lokalisierte Adsorption zu beschreiben, ist die Langmuir´sche Adsorptionstheorie (s. z. B. [Hil62/b]). Sie fußt auf der Definition eines "Gittergases", welches dadurch gekennzeichnet ist, daß die Substratoberfläche als starre Matrix mit N äquienergetischen Adsorptionsplätzen angesehen wird. (Diese müssen nicht zwingend mit der Anordnung der Substratatome übereinstimmen.) Die Gitterplätze sind statistisch (0 oder 1) besetzt, die Teilchen sind zunächst gleich verteilt.
Die Adsorbatphase kann mittels der Zustandssumme beschrieben werden. Sind so viele Adteilchen wie Adsorptionsplätze vorhanden, besteht diese aus der N-ten Potenz der Zustandssummen der einzelnen Teilchen Z = zN/N!. Sind weniger Adteilchen vorhanden, muß deren Verteilung berücksichtigt werden:
|
|
(A34) |
){kind=link}
Hat man es, wie in Metall-Adsorbatphasen, mit Teilchen zu tun, die untereinander wechselwirken, so müssen diese interatomaren Kräfte in die Überlegungen einbezogen werden. Daraus folgt die Möglichkeit, daß damit eventuell auftretende Phasenübergänge beschrieben werden können. Jedes Metall-Adteilchen besitzt, je nach Symmetrie, in der Oberfläche c nächste Nachbarn, mit denen es nun (attraktiv oder repulsiv) interagieren kann. (Auch weiterreichende Wechselwirkungen sind denkbar und werden in den verschiedenen Theorien unterschiedlich berücksichtigt.)
Für solche Systeme können in zwei- oder höherdimensionalen Fällen keine analytischen Lösungen gefunden werden, und man muß z. B. zur Anwendung von Näherungen übergehen. Zwei solche Näherungen, die häufig zum Einsatz kommen, sind die Bragg-Williams-Näherung (Bragg-Williams Approximation, BWA) und die Quasichemische Näherung (Quasi Chemical Approximation, QCA). Die BWA soll im Folgenden beschrieben werden.
3.5. Bragg-Williams-Näherung
Als ein wegen seiner mathematischen Einfachheit gut zu überschauendes Modell wurde zur Beschreibung der Adsorbat-Wechselwirkungen in Desorptionssystemen die angepaßte Bragg-Williams-Näherung gewählt. Diese beschreibt eigentlich (in ihrer ursprünglichen Form [BrW34/1]) Zweikomponentensysteme, kann aber auf Einkomponentensysteme, wie nicht (im Sinne einer Mischung) mit dem Substrat wechselwirkende Ad- bzw. Desorptionssysteme übertragen werden, die dann als Quasi-Zweikomponentensysteme angesehen werden, s. u.. Mit ihrer Hilfe läßt sich der Verlauf der systembestimmenden physikalischen Größen (wie Paar-Wechselwirkungsenergien, Bindungspotential) im Desorptionsexperiment relativ gut verstehen.
3.5.1. Definition des Systems
Da es sich bei der BWA "nur" um eine Näherung handelt, sollen zunächst die gleichen Einschränkungen gelten, die schon in
(All diese einschränkenden Voraussetzungen sind bei den in dieser Arbeit gewählten Systemen Cu; Ag; Au/Re(0001) recht gut erfüllt, zumindest dann, wenn der Bedeckungsgrad auf den Submonolagenbereich beschränkt wird. Nahe der Monolagenbedeckung können nämlich Wirkungen des misfits (z. B. -6,75 % beim Cu oder +5,4 % beim Ag) und einer eventuellen Besetzung der zweiten Lage zu Abweichungen führen.)
Nach der Gittergasnäherung (s. o.) existieren ˝ c QA N Adteilchenpaare AA auf der Oberfläche. Die Wechselwirkung eines Teilchens A mit dem restlichen "meanfield" ist ˝ c QA EAA, die einer Teilchenart folglich ˝ c N QA EAA. Die laterale Wechselwirkungsenergie des betrachteten Adsorbatteilchens mit seinen nächsten Nachbarn kann attraktiv (EAA < 0) oder repulsiv (EAA > 0) sein.
Wie schon eingangs erwähnt, beschreibt die BWA in ihrer ursprünglichen Form (dreidimensionale) binäre Legierungssysteme. Infolgedessen werden zwei Adteilchenarten A und B definiert, und es können sich Teilchenpaare AA, BB und AB bilden (N = NA + NB). Es gilt also (mit Q = Q A + Q B):
Bei der Bildung von NAB Paaren AB sind ˝ NAB AA- und BB- Paare verschwunden:
|
|
(A36) |
){kind=link}
Die bei der Berücksichtigung der Wechselwirkung AB auftretende Energiekorrektur beträgt:
|
|
(A37) |
){kind=link}
für ein Teilchen. Für das gesamte System ergibt sich ˝ c NAB DE (BWA) [Spa85/1].
3.5.2. Ableitung des chemischen Potentials
Um (eine) Desorptionsratengleichung(en) für die TD-Versuche zu erhalten, soll nun das chemische Potential μ des jeweiligen Adsorbatsystems errechnet werden. Mit dessen Hilfe läßt sich weiterhin die Phasengrenze eines eventuellen Phasenüberganges bestimmen, der die Desorption beeinflussen kann. Um zum chemischen Potential zu gelangen, geht man von der Definition der Zustandssumme aus, woraus sich wiederum die freie Energie des Systems berechnen läßt.
Durch die Berücksichtigung der Wechselwirkungen der nächsten Nachbarn wird ein zusätzlicher Term in der Zustandssumme
|
|
(A38) |
){kind=link}
Die atomare Zustandssumme z, die u. a. auch die Wechselwirkung mit dem Substrat beschreibt, soll zunächst zur Vereinfachung der Rechnungen abgetrennt werden. (Sie wird in
){kind=link}
|
|
(A39) |
){kind=link}
Übrig bleibt die BWA-Zustandssumme ZBWA, die die losgelöste Schicht aus wechselwirkenden Teilchen (A+B) beschreibt.
Zur Vereinfachung wird die totale Anzahl der Konfigurationen W eingeführt, die angibt, wie viele Möglichkeiten bestehen, Teilchen A und B auf N Plätzen anzuordnen:
|
|
(A40) |
){kind=link}
Mit
|
|
(A41) |
){kind=link}
Als nächstes soll die Freie Energie A des wechselwirkenden Systems berechnet werden. Durch Einsetzen von
|
|
(A42) |
){kind=link}
der sich gemäß (A = U - TS) in einen energetischen und einen entropischen Anteil zerlegen läßt. Für die innere Energie ergibt sich die Beziehung:
|
|
(A43) |
){kind=link}
und für die Entropie des Mischsystems:
|
|
(A44) |
){kind=link}
Führt man in (A42) den Bedeckungsgrad NA = QAN ein, so erhält man:
|
|
(A45) |
){kind=link}
Dieser Ausdruck stellt das System als aus Paaren AA, BB und AB aufgebaut dar. Der Prozeß der Durchmischung, bei dem durch die Wirkung von DE Paare AB gebildet werden, läßt sich beschreiben, indem in
|
|
(A46) |
){kind=link}
Die Komponenten A und B sollten sich mit negativem EWW mischen und mit positivem EWW voneinander separieren.
Wie einleitend erwähnt, wird bei der Anwendung der BWA auf Adsorbatsysteme, bei denen jeweils nur eine Adteilchenart vorhanden ist, NA mit der Anzahl der Adteilchen identifiziert und NB mit der Zahl der leeren Gitterplätze N0 (somit gilt Q = QA). N0 ist damit eine Quasikomponente (eine reine Rechengröße, aber keine stoffliche Komponente) und das System NAN0 ein Quasizweikomponentensystem. Damit ist sowohl die energetische Wechselwirkung zweier leerer Gitterplätze miteinander Null (EBB ® E00 = 0), als auch die zwischen leeren und mit Adteilchen besetzten Gitterplätzen (EAB ® EA0 = 0).
Eine ideale Mischung von Adteilchen und leeren Plätzen entspricht der idealen Verteilung der Teilchen in Form eines 2D-Gases auf äquivalenten Gitterplätzen. Der Prozeß der Entmischung steht für die zusätzliche Ausbildung von Adsorbatinseln (kondensierte Teilchen) auf Grund attraktiver Wechselwirkungen, d. h. für das Auftreten einer zweiten Phase bzw. die Ausbildung eines Phasengleichgewichtes. Dies ist mit EWW > 0 der Fall. Mit EWW < 0 kommt es durch repulsive Wechselwirkungen der Adteilchen untereinander dazu, daß die Teilchen einen möglichst weiten Abstand voneinander einnehmen, es bildet sich eine 2D-Gasphase (oder speziell geordnete Phasen, in denen die Teilchen weit von einander getrennt sind).
Allerdings kann man EWW auch als eine reine Paar-Wechselwirkungsenergie auffassen. Nach
| EWW = -EAA | (A47) |
){kind=link}
Werden bereits in
Zum chemischen Potential μ für das zweidimensionale System gelangt man, wegen
|
|
(A48) |
){kind=link}
(j = Oberflächendruck, a = Oberfläche) mit p dV = 0 (für Oberflächen) durch Differenzieren von (A46) nach N bzw. Q:
|
|
(A49) |
){kind=link}
Gemäß
|
|
(A50) |
Für die partielle Entropie als Konfigurationsteil gilt:
|
|
(A51) |
In
Für EWW = 0 ergibt sich der Langmuir´sche Fall und für EWW < 0 (repulsive Wechselwirkungen) sind keine Extrempunkte erkennbar. Bei repulsiver Wechselwirkung wird also gar keine Gleichgewichtsphase 2D-Gas/2D-Kondensat ausgebildet.
Mit EWW > 0 (attraktive Wechselwirkungen) hat das chemische Potential zwei Extremstellen. Im Bereich zwischen den Extrema stehen 2D-Gas- und Inselteilchen im Gleichgewicht. Dieser Bereich vergrößert sich mit zunehmender positiver EWW (bzw. Koordinationszahl c). Das Auftreten von Inseln ist also um so mehr begünstigt, je höher die attraktive Wechselwirkung zwischen den Teilchen ist.
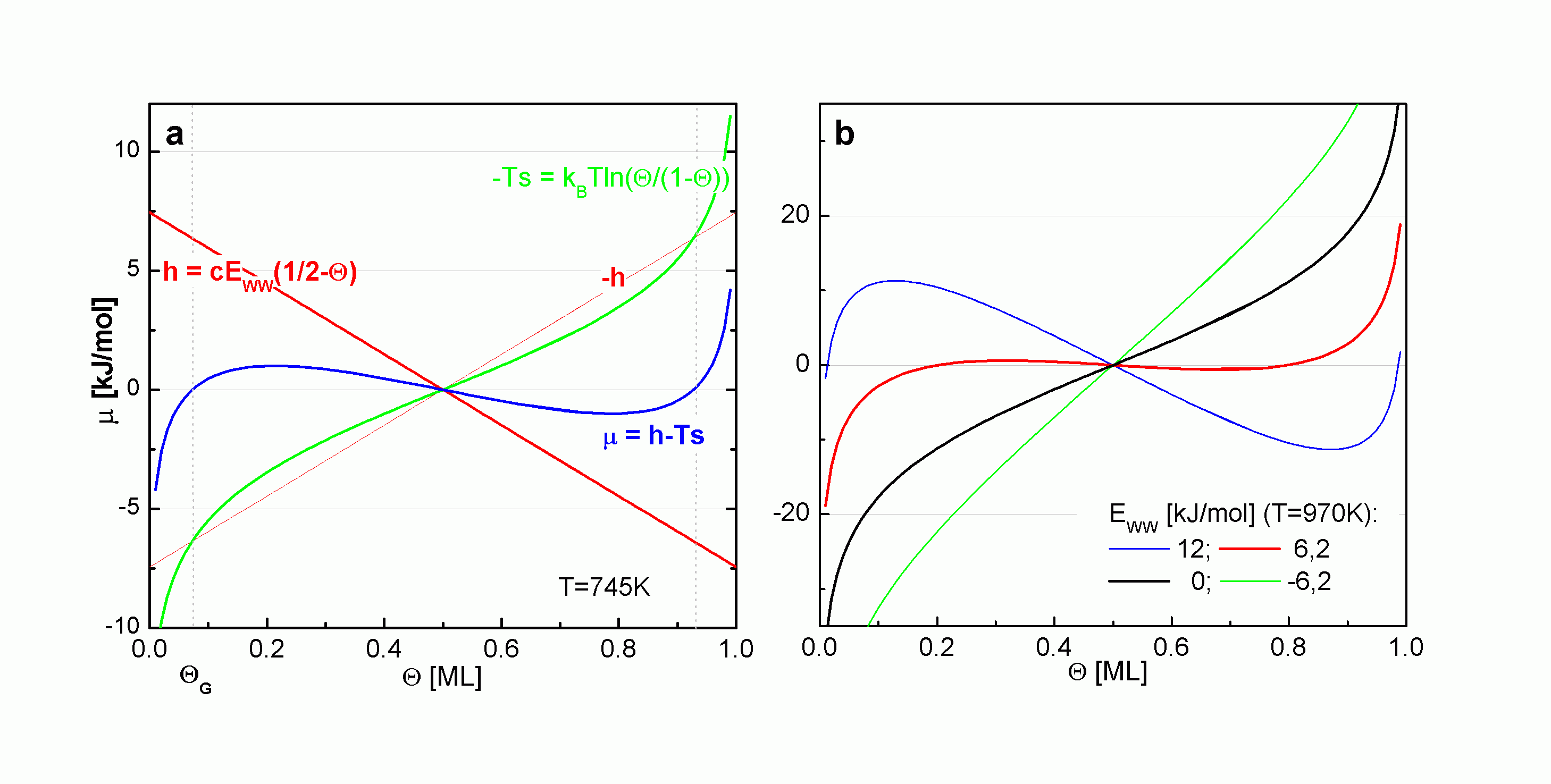
| |
| Abbildung A10 a: Zusammensetzung des chemischen Potentials aus enthalpischen und entropischen Anteilen, b: chemisches Potential für verschiedene EWW |
|
Im gesamten Zweiphasengebiet besteht das Gleichgewicht zwischen Bildung und Auflösung von Inseln (dem Entmischungs- und Mischungsvorgang), was daraus resultiert, daß der energetische Beitrag zur Ablösung der Adteilchen von Inseln vom entropischen Beitrag kompensiert wird. Es gilt also mBWA = 0. Geht man von dieser Aussage aus, so muß festgestellt werden, daß der Verlauf des chemischen Potentials in dem dazugehörigen Bedeckungsgradbereich mit der "van-der-Waals-Schleife" durch die BWA nicht richtig beschrieben wird. In der Eingangsvoraussetzung zur Beschreibung eines Molekularfeldes wird die Gleichverteilung der Teilchen gefordert. Die Ausbildung von Inseln wäre somit gar nicht möglich. Summiert man jedoch über alle mögliche Anordnungen der Teilchen auf der Oberfläche (bzw. mit N ® ¥), so erhält man im Bereich zwischen den Nullstellen einen konstanten Verlauf.
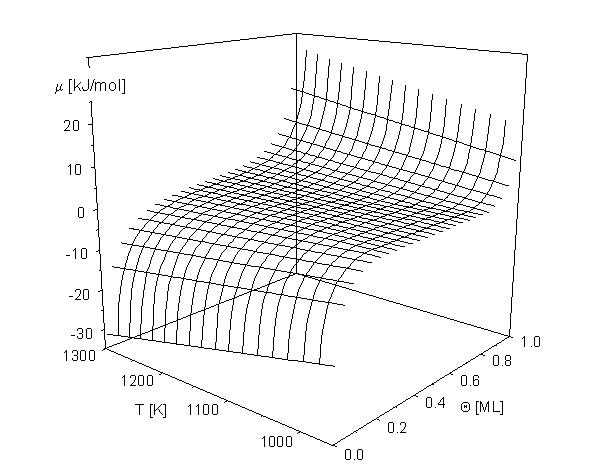
| |
| Abbildung A11 chemische Potentialfläche für EWW = 6,2 kJ/mol. |
|
Auf graphischem Wege erhält man durch die Anwendung der Maxwell´schen Konstruktion [KoB85/1, PaB87/1] eine dreigeteilte Kurve mit dem konstanten Verlauf des effektiven chemischen Potentials im Bereich des Phasengleichgewichtes, wenn sich die Flächen zwischen der mBWA- und der m = 0-Linie zu Null ergänzen [Zhd85/1]. Der Einfluß der Wechselwirkungsenergie und der Temperatur auf das chemische Potential wird in
3.5.3. BWA-Phasendiagramm
Die Temperaturabhängigkeit (der Lage der Nullstellen) des chemischen Potentials kommt in der Form der Phasengrenze zum Ausdruck. Es ergibt sich bei Q = 0,5 ML ein spiegelsymmetrischer Verlauf dieser Phasengrenze mit Q (
Beim Einsetzen des Desorptionsvorganges bei der Grenztemperatur TG bilden sich die Inseln erst ab dem Grenzbedeckungsgrad QG und bei Erreichen der kritischen Temperatur existieren nur noch homogen verteilte (2D-Gas-) Teilchen. Bei Raumtemperatur liegt fast im gesamten Q -Bereich das Zweiphasengebiet vor.
Die Bedingung mBWA = 0 für die Koexistenz von 2D-Gas- und Kondensatphase, wird (ab) dann erfüllt, wenn gilt: h = Ts. Die Phasengrenze berechnet sich mit
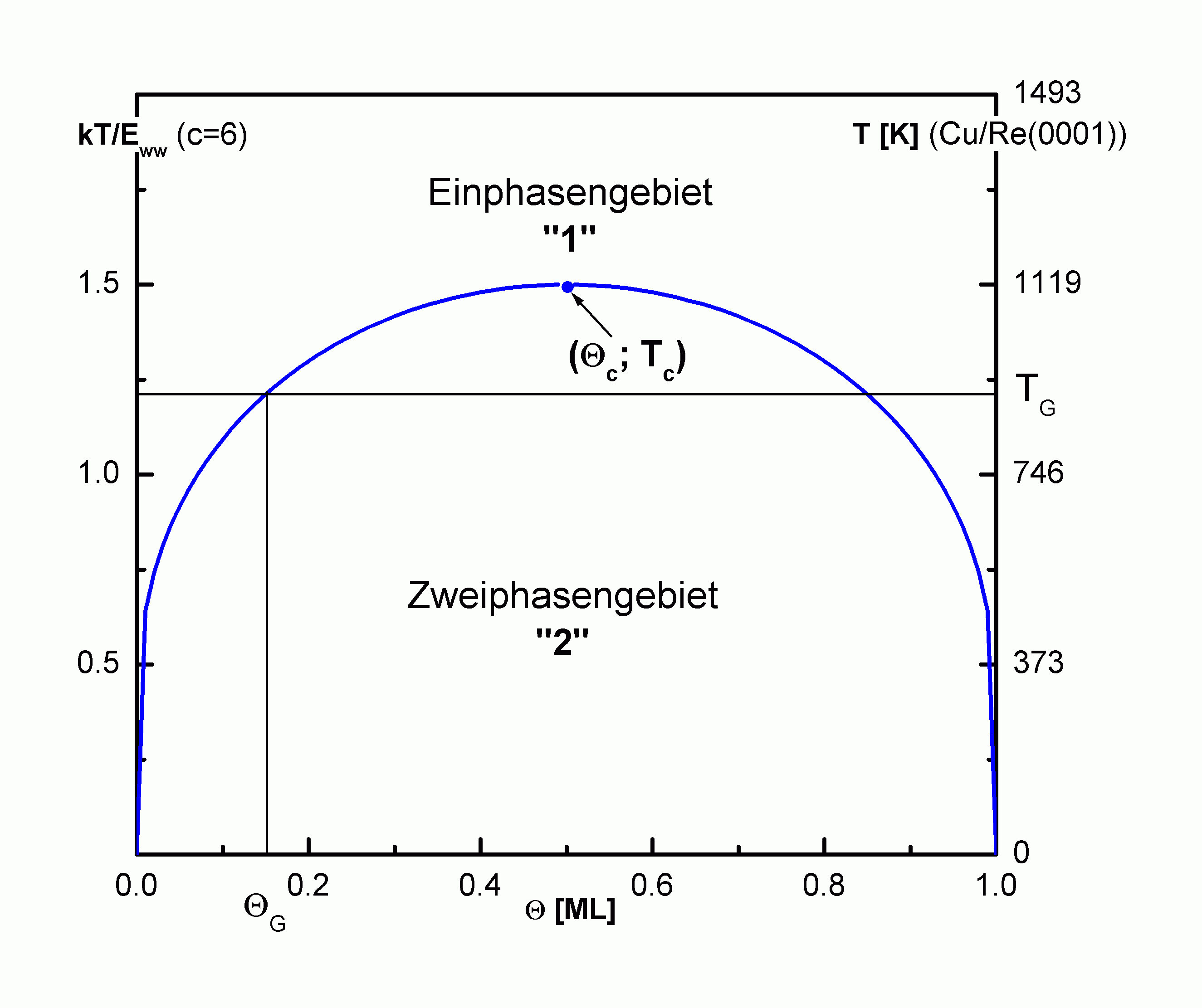
| |
|
Abbildung A12
Phasendiagramm für ein paarweise attraktiv wechselwirkendes Adsorbatsystems nach BWA mit c =6 (Temperaturskala auf der rechten Seite für Cu/Re(0001) mit EWW = 6,2 kJ/mol, s. |
|
|
|
(A52) |
){kind=link}
Aus dieser Koexistenzbedingung läßt sich EWW bestimmen, indem man Q ® 1/2 und T = TC setzt (TC = kritische Temperatur des Phasenübergangs) [PaB87/1, NSH84/1, Nag85/1]. Es ergibt sich der allgemeine Zusammenhang:
|
|
(A53) |
){kind=link}
Im Phasendiagramm eines paarweise attraktiv wechselwirkenden Adsorbatsystems besteht bei kleinen Bedeckungen nur die 2D-Gasphase, die von einer Zweiphasen-Koexistenzregion von einer 2D-(eher)-flüssigen Phase bei hohen Bedeckungen getrennt wird (
3.5.4. Desorptionsratengleichungen
Wie sich in
| mBWA = - m1 + m2 | (A54) |
){kind=link}
Nur im Gebiet 1 kann Q einer konkreten Phase zugeordnet werden (Q1 = Q2D-Gas (bzw. Q1 = Q2D-Kond. )) und μ ist abhängig von Q, im Gegensatz zu Gebiet 2 (Q2 = Q2D-Kond. + Q2D-Gas ), wo μ unabhängig von Q ist:
|
|
(A55) |
){kind=link}
|
|
(A56) |
){kind=link}
Geht man von einer Gleichung für ZBWA äquivalent zu
|
|
(A57) |
){kind=link}
Nach
|
|
(A58) |
Wie schon bei
|
|
(A59) |
){kind=link}
Um einen Ausdruck für die Desorptionsrate zu erhalten, geht Kreuzer vom Modell eines Massenflusses aus, der aus dem Vorhandensein einer (3D-) Adsorbatgasphase resultiert, welche mit dem Adsorbat im "Quasigleichgewicht" steht [Kre91/b, Kre92/1]. Diesen Massenfluß kann er über die translatorische 3D-Zustandssumme und das chemische Potential der 3D-Gasphase ausdrücken. Unter den Voraussetzungen, daß 1.) im Quasigleichgewicht dieses chemische Potential dem der Adsorbatphase gleich sein muß und somit gleichgesetzt werden kann sowie daß 2.) die desorbierenden Teilchen ausschließlich aus der 2D-Gasphase stammen, gelangt er über einige Näherungen zu folgender Ratengleichung:
|
|
(A60) |
){kind=link}
Darin ist die gesamte Q - Abhängigkeit in μBWA enthalten [PaB87/1, Kre91/b, Nag86/1, NaH88/1]. Der Vorfaktor dieser Exponentialgleichung besteht in diesem Fall (die ausschließliche Desorption von 2D-Gasteilchen wurde vorausgesetzt) einzig aus der Frequenz ν, die aus der Zustandssumme der Schwingung der Adteilchen gegen die Substratoberfläche stammt.
Die Desorptionsrate kann für die beiden Bereiche 1 und 2 formuliert werden, indem man
|
|
(A61) |
){kind=link}
|
|
(A62) |
){kind=link}
Diese Darstellung entspricht der Polanyi-Wigner-Gleichung mit einem Korrekturterm. Hier ist die makroskopische Meßgröße Desorptionsenergie durch die mikroskopischen Größen, die systembestimmenden Größen Oberflächenpotential in z-Richtung, Vz,0 und die laterale Wechselwirkungsenergie EWW ersetzt bzw. ausgedrückt worden.
Mit den Gleichungen (A61) und (A62) lassen sich sowohl der isotherme Verlauf der Desorptionsenergie als auch der Verlauf der Desorptionsrate als Funktion des Bedeckungsgrades beschreiben.
- Die Desorptionsenergie steigt an, solange Teilchen aus dem Einphasengebiet desorbieren. Die Desorption aus dem Zweiphasengebiet ist durch eine konstante Desorptionsenergie gekennzeichnet [PaK89/1]. Damit hängt der Verlauf der Desorptionsenergie davon ab, in welchen Temperatur- und Bedeckungsgradbereichen das Desorptionsexperiment durchgeführt wird.
- Die isotherme Desorptionsrate ist ebenfalls konstant, solange aus dem Zweiphasengebiet desorbiert wird. Sie ist deshalb nicht bedeckungsgradabhängig, weil hier die aus der 2D-Gasphase bei der Desorption verloren gehenden Teilchen solange sofort durch Teilchen, die aus den 2D-Inseln stammen, ersetzt werden, solange diese Inseln existieren.
Die Desorptionsrate aus dem Einphasengebiet steigt hier exponentiell mit dem Bedeckungsgrad an. Dies ist insbesondere aus einer linearen Steigung in diesem Q-Bereich in den "order plots" zu entnehmen. Der Übergang einer solchen Isothermen zum linearen Bereich kennzeichnet die Phasengrenze.