|
|
|
Kapitel C4 | Abschnitt C - Münzmetallschichten auf Rhenium |
|
|
|
Kapitel C6 | Kapitel C5 - Der Phasenübergang der 2D-Verdampfung |
5. Der Phasenübergang der 2D-Verdampfung
Wie in
Wie bereits in den Kap.
Da die Methoden zur Gewinnung der kritischen Größen aus den TD-Spektren nicht trivial sind, ist es von großem Nutzen, die Meßdaten mehrerer Systeme gemeinsam auszuwerten. Aus diesem Grund soll im folgenden Kapitel für die drei untersuchten Edelmetall-auf-Rhenium-Systeme vergleichend dargestellt werden, wie Messung und Auswertung des Phasenübergangs der zweidimensionalen Verdampfung durchgeführt wurden. Insbesondere werden folgende Schwerpunkte behandelt:
- Darstellung und Vergleich der Methoden zur Bestimmung der Phasengrenze
- Messung der kritischen Größen, Berechnung der Adsorbat-Wechselwirkungsenergie EWW
- Simulation des Verlaufs von EWW und Vergleich mit Edes
- Ermittlung von realistischen Wechselwirkungsenergien EWW (real), VZ,0
- Einfluß des ps-cp-Übergangs auf VZ,0
5.1. Messung der Phasengrenze
Der Verlauf der Phasengrenze kann auf unterschiedliche Art und Weise gemessen werden. Einerseits ist es möglich, durch Untersuchung der Morphologie des Films Rückschlüsse auf das Vorhandensein von Adteilchen in nur dem einen oder in beiden 2D-Bindungszuständen zu ziehen. Solche Untersuchungen können z. B. mit STM-Messungen (Au/Ru(0001) [GGK93/1], Ag/Pt(111) [RBK94/1], Ag/W(110) [JMN90/1]) oder auch DF-Messungen (Ag/Ru(0001) [NSW95/1], Cu;Ag;Au/W(110) [KoB84/2, KoB85/1], Cu;Ag/Mo(110) [Kol87/1, Kol90/1]) vorgenommen werden.
Die Phasengrenze wird aber auch durch ihre Wirkung auf gleichzeitig ablaufende Prozesse wie die Thermodesorption bestimmt. Literaturbekannt sind unterschiedliche Methoden, aus TD-Serien die Phasengrenze zu bestimmen, etwa über Desorptionsisothermen oder Arrhenius-plots, s.
Es wurde in den vorangegangenen Kapiteln schon ausgeführt, daß die Desorption von Cu, Ag und Au auf der Re(0001)-Oberfläche in unterschiedlichen Temperaturbereichen erfolgt. Durch Variation der Heizrate gelingt es, diesen Temperaturbereich um bis zu 100 K zu verschieben. Beim Cu gelingt es sogar, die Desorption einmal aus dem Ein-, dann aus dem Zweiphasengebiet ablaufen zu lassen.

Um die Phasengrenze zu bestimmen, wurden für das Cu zwei TD-Serien LT (b = 7,7 K/s) und HT (b = 31,8 K/s), für das Ag zwei TD-Serien LT (b = 4,1 K/s) und HT (b = 26,9 K/s) sowie für das Au eine Serie LT (3,68 K/s) zunächst mit der "Flankenmethode" untersucht. Die Bestimmung der Phasengrenze war jedoch, bedingt durch die Spektrenform, nur bei der Cu-LT- und der Ag-HT-TD-Serie möglich, s.
Daraufhin werden die Spektren von der Hochtemperaturseite her integriert, um den Verlauf der TD-Pfade (vgl.
In einem solchen Phasendiagramm kann die Phasengrenzlinie nur bis zu dem Punkt bestimmt werden, an dem die Desorptionspfade die Phasengrenze durchschneiden. Um die kritische Temperatur TC und den kritischen Bedeckungsgrad QC zu erhalten, wird die Phasengrenze im Rahmen der Bragg-Williams-Näherung (
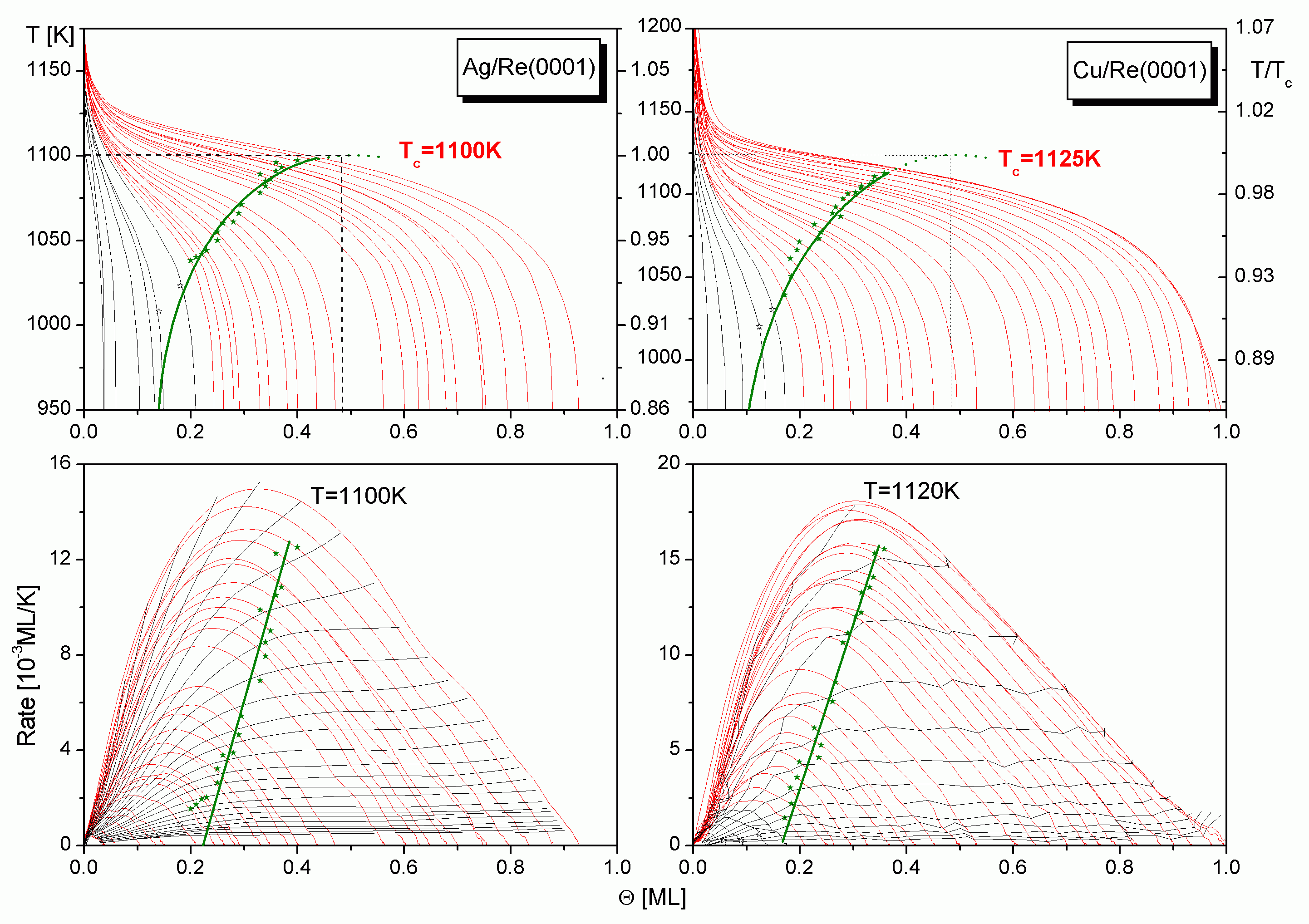
In
Die simulierte Phasengrenze wurde von hier in die TD-Fläche sowie in die (Q; R)- und die (T; R)-Ebenen projiziert. Der Verlauf der (Q; R)-Projektion der BWA-Phasengrenze ist in einem großen Teil des ansteigenden Astes ebenso linear wie der der gemessenen Grenzlinie.
Werden die Wertepaare (TG; QG) in die layer plots der Systeme, also in die Desorptionsebene (Q; R)-Ebene eingetragen (Projektion in die Ratenebene (Q; R)), erhält man in einem weiten Bereich, nämlich zwischen 0,17 ML und 0,35 ML für das Kupfer sowie 0,22 ML und 0,35 ML für das Silber, einen linearen Verlauf der Phasengrenze, s.
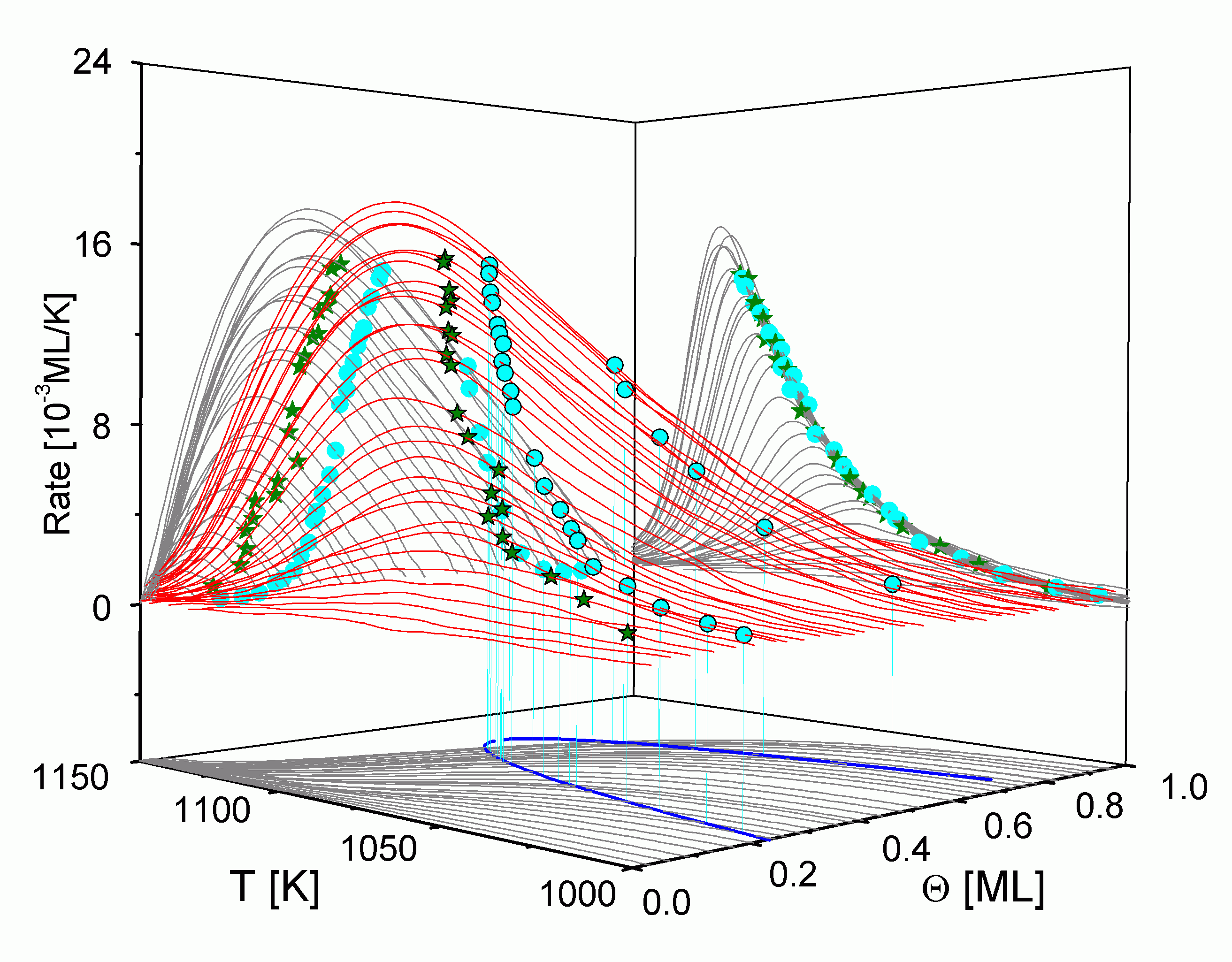
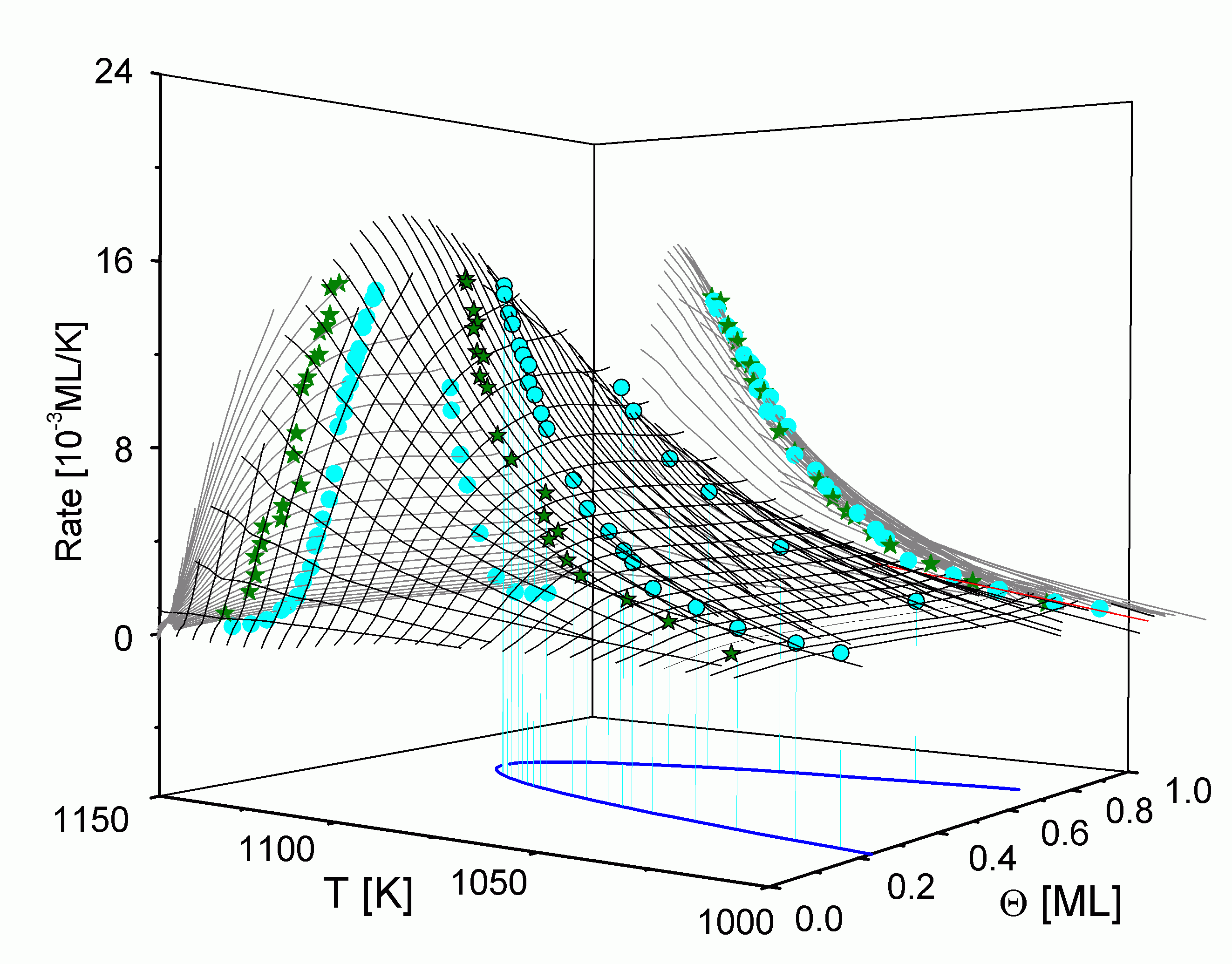
Werden in
Die Isothermen zeigen über einen weiten Bereich einen horizontalen Verlauf, wie er auch für andere Systeme gefunden wird [PaB87/1, NaH87/1, KrP88/1, Kre90/1, Kre90/2]. Ausgehend von der Vorstellung, daß die Teilchen nur aus der 2D-Gas-Phase heraus desorbieren, bleibt die Desorptionsordnung solange Null, wie Teilchen aus der kondensierten Phase mit einer hohen Geschwindigkeit in 2D-Gasteilchen überführt werden, also das Phasengleichgewicht eingestellt bleibt. Im Bereich davor und danach ist ein starker Anstieg der Isothermen zu verzeichnen, der der Desorption aus dem Einphasengebiet zuzuordnen ist [EGC86/1]. Die Übergangspunkte von diesem starken Anstieg zum horizontalen Verlauf kennzeichnen also die Phasengrenze.
Die Desorptionsisothermen sind auch in
Für den Teil der Phasengrenze mit Q > 0,5 ML lassen sich leider keine genauen Aussagen treffen, da dieser Bereich durch das Desorptionsexperiment nicht erfaßt wird. Allerdings liegt der Übergang vermutlich außerhalb des Desorptionsbereiches, da kein erneuter Anstieg der Isothermen zu verzeichnen ist. (Lediglich ab 0,9 ML kann ein geringer Anstieg vermutet werden.)
Als ein für die durchgeführte Messung charakteristischer Wert wurde der tiefste Bedeckungsgrad Q G0 bestimmt, bei welchem die Phasengrenze mittels TD-Spektroskopie betrachtetet werden kann. Das ist der Anfangsbedeckungsgrad, bis zu dem die TD-Pfade ausschließlich im Einphasengebiet liegen bzw. überhaupt noch nicht in der gemeinsamen Anstiegsflanke laufen.
Diese Grenzbedeckung kann aus der Zustandsebene der Systeme nur relativ ungenau bestimmt werden. Durch den linearen Verlauf von QG in der Ratenebene (Q; R) läßt sich hier QG0 jedoch sehr einfach bestimmen. Extrapoliert man nämlich den linearen Teil der Phasengrenze auf die Q-Achse, so erhält man mit großer Genauigkeit den Wert von QG0. Aber auch aus den Isosteren aus
| Cu | Ag | |
TC [K] |
1125 | 1100 |
QC [ML] |
0,48 | 0,48 |
| QG0 [ML] | 0,16 | 0,22 |
Vergleichswerte anderer Systeme sind der Literaturtabelle im Anhang zu entnehmen. Die Literaturwerte der kritischen Temperaturen liegen für Kupfer im selben Temperaturbereich, soweit sie durch TDS-Methoden bestimmt wurden. Die aus DF-Messungen erhaltenen Werte sind demgegenüber ca. 80 bis 150 K geringer. Für Silber liegen die Tc-Werte, die aus DF-Messungen und aus TDS-Methoden gewonnenen wurden, im selben Bereich. Das kann daher rühren, daß Ag-TD-Serien untersucht wurden, die in einem zu niedrigen Temperaturbereich lagen. (Bei der Flankenmethode wirkt sich dies z. B. als methodischer Fehler aus, da die TD-Spektren die gemeinsame Anstiegsflanke gar nicht auf Grund des Phasenüberganges, sondern wegen der Leerung des TD-Zustandes verlassen).
Parallelen zum 3D-Schmelzvorgang ergeben sich insofern, als daß man gar nicht genau festlegen kann, ob die 2D-freibeweglichen Teilchen einen 2D-gasförmigen oder 2D-flüssigem Aggregatzustand besitzen. Auch letzterer kann mit der BWA beschrieben werden, und man erhält ähnliche Zusammenhänge wie in
| Cu | Ag | Au | |
| ľ Sp. [K] ( |
1018 | 926 | 1002 |
| TC [K] | 1125 | 1100 | (1137) |
Die Q C-Literaturwerte für die Vergleichssysteme (Cu;Ag;Au/Mo(110) u. ä., s. Literaturtabelle) liegen durchweg bei niedrigeren Bedeckungsgraden (0,2 bis 0,35 ML). Ursache dafür ist vermutlich das hier gewählte methodische Vorgehen. Während bei der durchgeführten Auswertung der symmetrische Verlauf der Phasengrenze (laut BWA) angenommen wurde, wird bei anderen Autoren QC entweder durch andere Näherungen (z. B. QCA) bestimmt oder sogar gemessen.
Aus den TC-Werten kann nunmehr nach
| Cu | Ag | ||
| EWW [kJ/mol] | BWA | 6,23 | 6,09 |
| QCA | 7,58 | 7,41 |
5.2. Simulation des Verlaufes der Desorptionswärme
Die Gleichungen
Der Übergang zwischen den beiden Gleichungen ist durch die Phasengrenze in der Zustandsebene des Systems (Q; T)
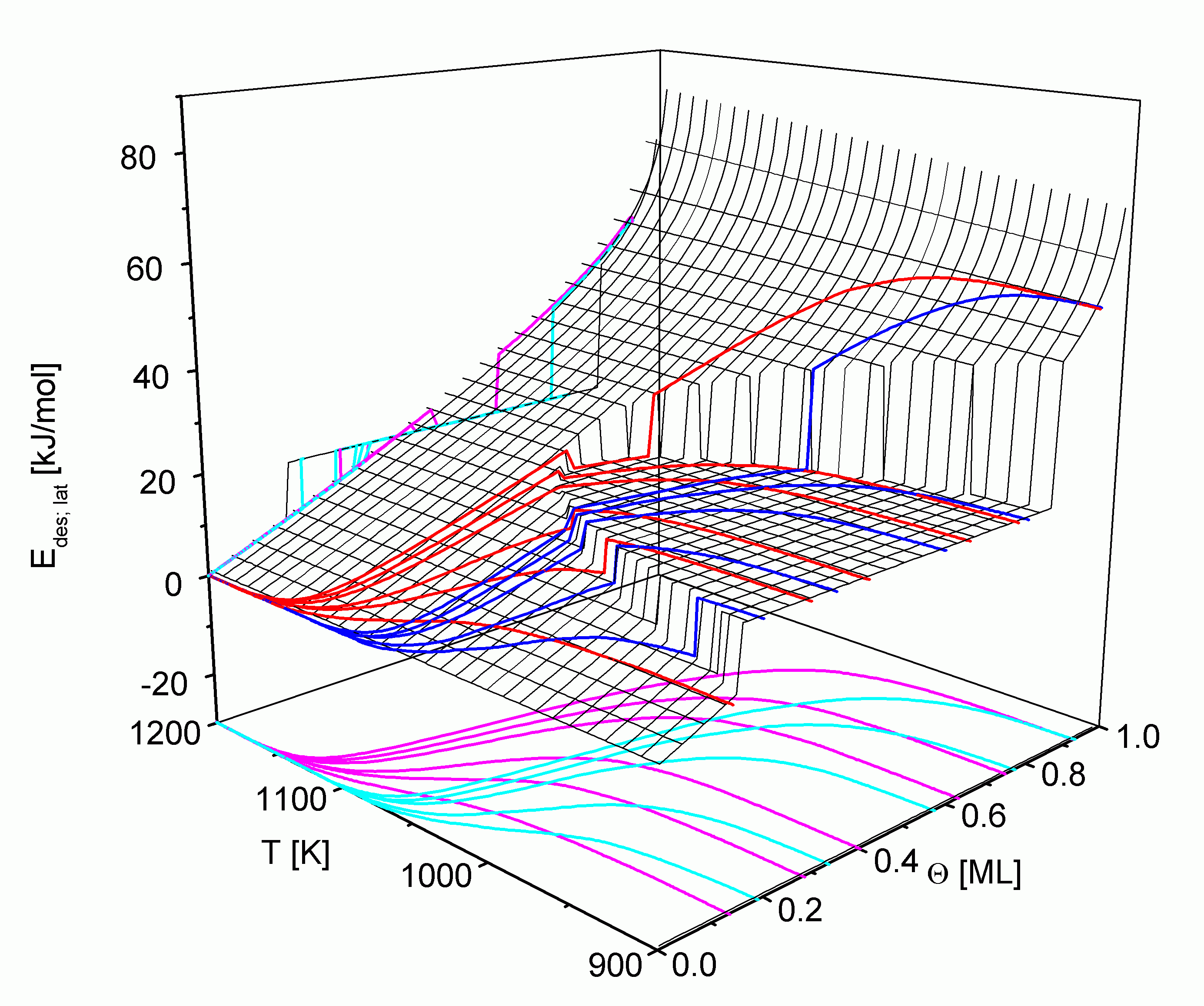

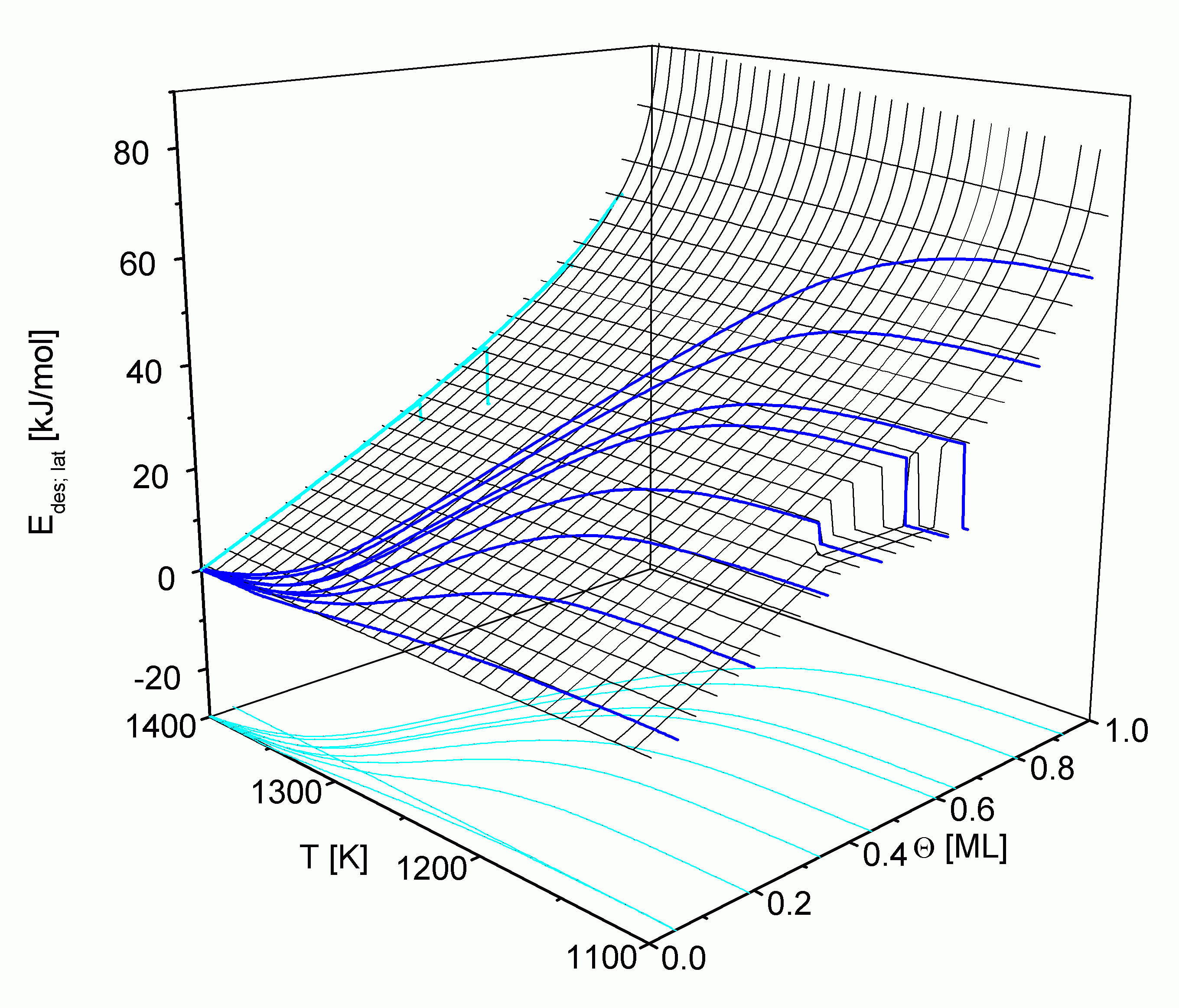
Dies wird in
Aus der Projektion in die Zustandsebene kann man wiederum die relative Lage der TD-Pfade entnehmen. Die zur T-Achse parallelen Abschnitte liegen außerhalb des jeweiligen Desorptionsbereiches.
Es ist jeweils das Gebiet dargestellt, in dem sich die Desorption über die LT- und die HT-TD-Pfade vollzieht. Erwartungsgemäß ergeben sich für die Energiefläche wiederum zwei Bereiche. Im Einphasengebiet 1 steigt Edes,lat hauptsächlich wegen der vermehrten attraktiven Wechselwirkungen bei hohen Bedeckungsgraden aber auch durch die Wirkung des Konfigurationsterms in
Der zweite Bereich liegt innerhalb der Phasengrenze und repräsentiert damit das Gebiet 2 des Phasengleichgewichtes. Er zeichnet sich durch einen horizontalen Verlauf von Edes,lat aus, der unabhängig vom Bedeckungsgrad ist und um so höher liegt, je größer EWW ist.
Da sich die kritischen Temperaturen der drei Metalle nur wenig voneinander unterscheiden, liegt das Zweiphasengebiet in allen drei Fällen bei etwa gleichen Temperaturen. Der Desorptionsbereich von Cu, Ag und Au unterscheidet sich jedoch merklich. Daraus resultiert, daß sich Desorptions- und Zweiphasenbereich beim Silber stark, beim Kupfer für die LT-Pfade ebenfalls stark und für die HT-Pfade schwach sowie beim Gold gar nicht überschneiden. Für das Kupfer-System sind zusätzlich drei LT-Pfade mit 1 ML < Q 0 < 2 ML eingezeichnet, die das Zweiphasengebiet ebenfalls kaum tangieren.
Aus der Projektion in die (Q; E)-Ebene aus
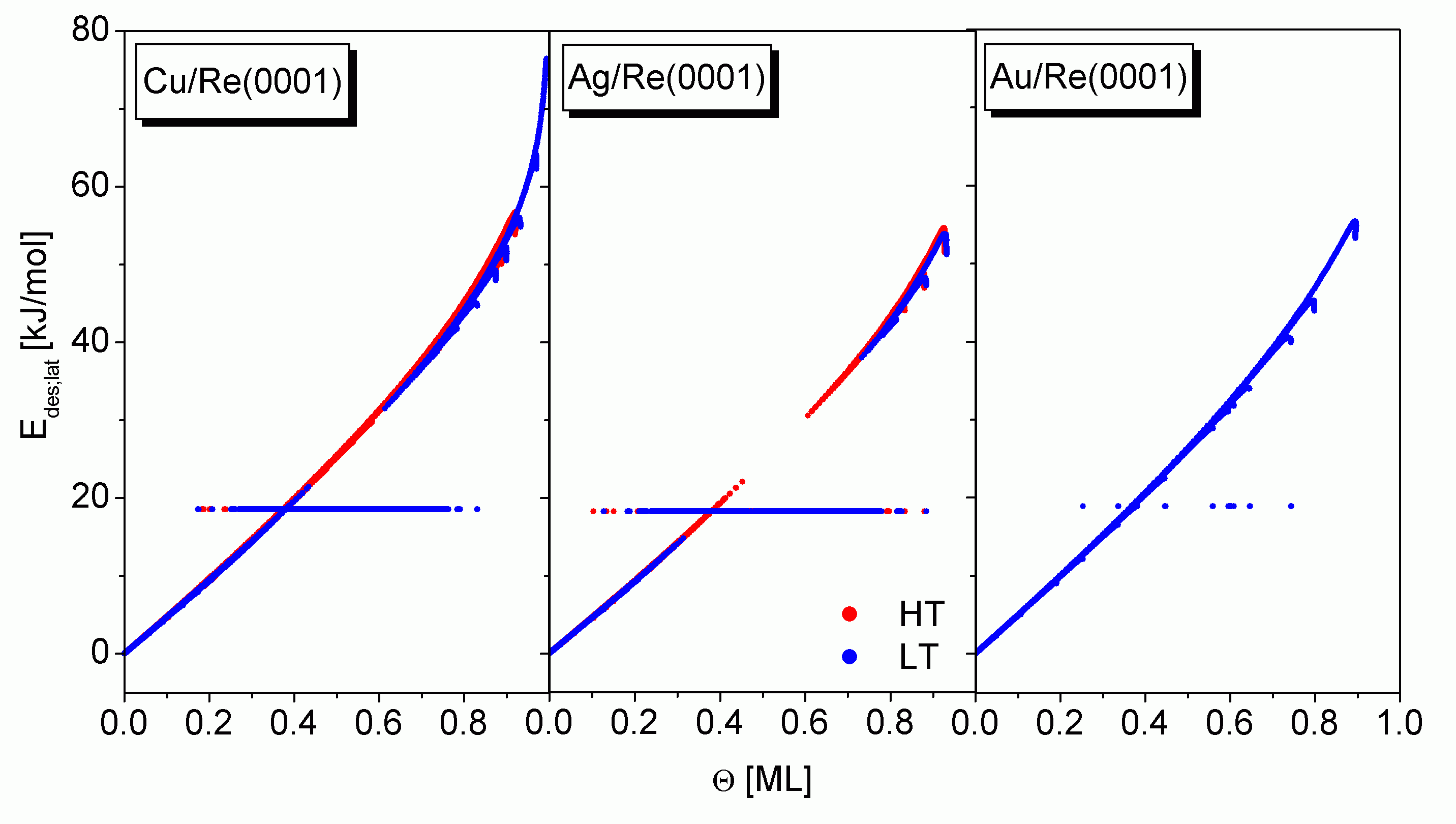
Um den Q-Verlauf der gemessenen Desorptionsenergie simulieren zu können, müssen die Erkenntnisse der bisherigen Betrachtungen an die Form der Auswertung der TDS adaptiert werden. Dies ist bei den Gleichungen
Überträgt man dieses Verfahren auf den vorliegenden Fall, so besteht der erste Schritt darin, die Desorptionstrajektorien in die Zustandsebene des Systems einzutragen, wie es in
Um ein Resultat zu erhalten, das sich mit den Meßergebnissen vergleichen läßt, reicht dies allerdings nicht aus. In
Zur Bestimmung des Energieverlaufes mit dem Bedeckungsgrad, der zur Bauerschen Auswertung äquivalent ist, muß über die Energie-Pfade Edes,lat(Q, T) aus
In
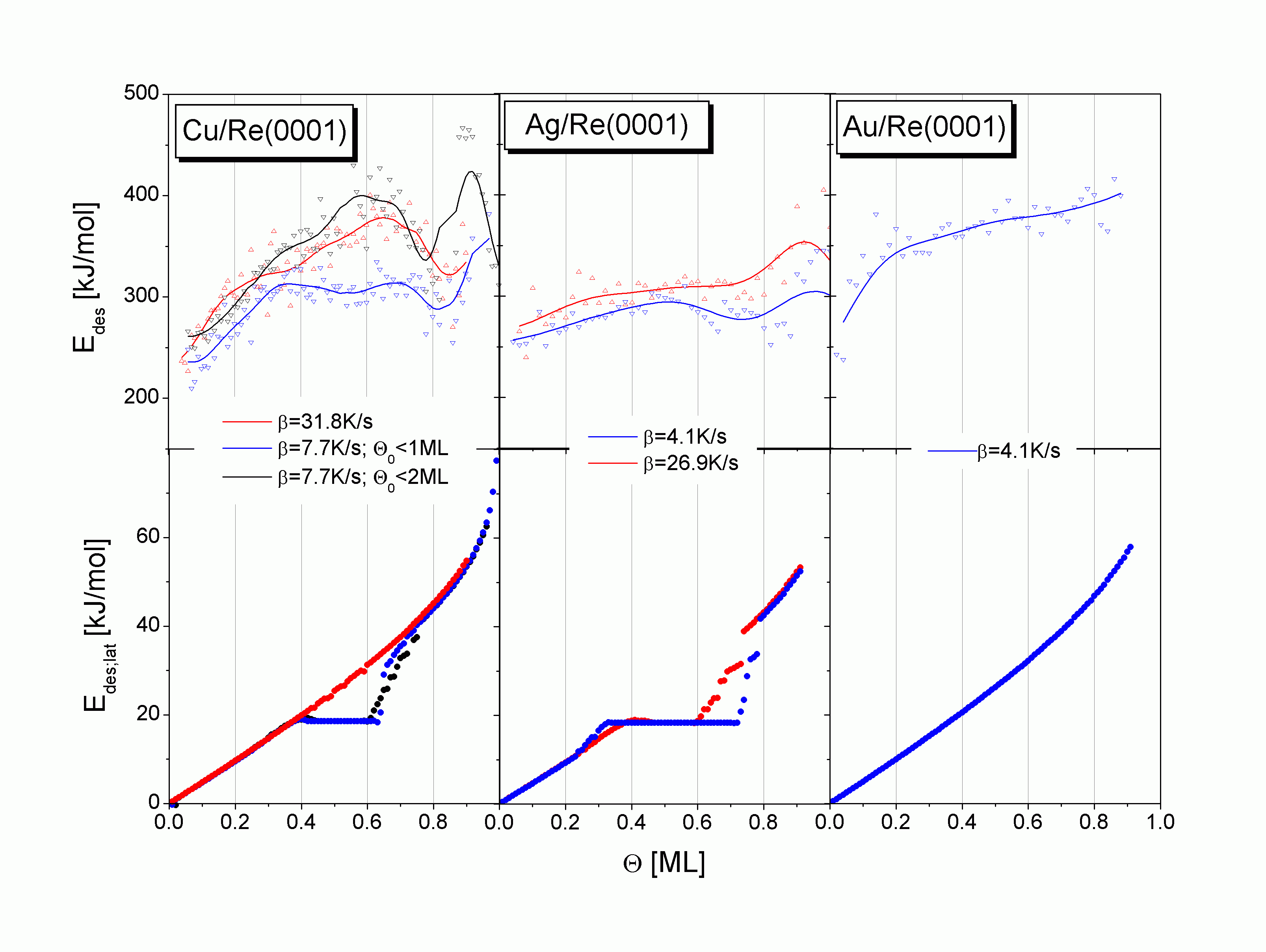
| |
| Abbildung C46 oben: Desorptionsenergie der Systeme Cu;Ag;Au/Re(0001), unten: Simulation des tempe-raturgemittelten lateralen Beitrages zur Desorptionsenergie. |
|
Erwartungsgemäß ergibt sich in den Simulationen für den Bereich bis 0,3 ML etwa der gleiche Kurvenverlauf für alle drei betrachteten Systeme. Er entspricht in allen Fällen einer Desorption erster Ordnung aus dem Einphasengebiet. Die Steigung der entsprechenden Kurven ist zur lateralen Wechselwirkungsenergie EWW der Adteilchen proportional und unterscheidet sich deshalb nur marginal. Der Verlauf entspricht dem Anstieg der gemessenen Edes-Kurven.
Der Bedeckungsgradbereich von 0,3 ML bis 0,8 ML ist durch einen stark voneinander abweichenden Verlauf der Kurven gekennzeichnet. Die aus den LT-TD-Pfaden des Cu erhaltenen Kurven sind von 0,4 bis etwa 0,6 ML konstant und steigen dann um etwa 45 kJ/mol an. Dieser Kurvenverlauf spiegelt das Wechseln der Desorptionsordnung von 1 nach 0 und wieder nach 1 wider, also den Übergang vom Ein- ins Zweiphasen- und wieder ins Einphasengebiet des zweidimensionalen Systems. Die Kurve der HT-Pfade zeigt in diesem Q-Bereich einen relativ konstanten Anstieg (n = 1, Einphasengebiet). Dabei besteht die größte Abweichung zwischen HT- und LT-Pfaden der Simulationskurven bei 0,6 ML mit etwa 25 kJ/mol. Der größte Unterschied zwischen LT- und HT-Pfaden der Meßwerte beträgt dagegen (ebenfalls bei 0,6 ML) etwa 60 kJ/mol.
Die in
Edes,lat von Ag ist sowohl für LT-Kurven (0,3 bis 0,7 ML) als auch für HT-Kurven (0,4 bis 0,6 ML) der Simulation durch einen horizontalen Verlauf gekennzeichnet. Dieses Verhalten findet sich, wie o. a., in ähnlicher Weise in den Cu-LT-Pfaden wieder und reproduziert sehr schön den Verlauf der experimentell bestimmten Desorptionsenergie des Silbers. Dabei fällt auf, daß es sowohl in der Simulation als auch in den Meßwerten der HT-Kurven etwa 0,1 ML früher zu einem Anstieg der Energie kommt als bei den LT-Kurven, was auf die Verschiebung des Temperaturbereiches der Desorption in Richtung Einphasengebiet (zu höheren Temperaturen) hinweist. Die Gesamtsteigerung der Energie liegt in diesem Gebiet für den Simulationsverlauf und ebenso für die experimentelle Desorptionsenergie bei 20 kJ/mol. Von Payne et al. konnten auf anderem Wege für das System Ag/Mo(110) ein äquivalenter Energieverlauf gefunden werden [PaK89/1].
Beim Gold sind die Simulations- und Meßkurven durch einen annähernd konstanten Anstieg gekennzeichnet. Ursache ist die Desorption aus dem Einphasengebiet. Die Gesamtsteigerung der Energie in diesem Gebiet liegt für die Simulationskurve bei 35 kJ/mol und die Desorptionsenergiekurve bei 40 kJ/mol. Beide stimmen somit gut überein.
Der Bereich ab 0,8 ML ist, wie bereits der Anfangsbereich, durch ein Anwachsen der Energie gekennzeichnet, das wiederum mit der Desorption aus dem Einphasengebiet erklärt werden kann. Allerdings ist der Funktionsverlauf hier nicht mehr linear. Dies hängt mit der Wirkung des Entropieterms in
Bei den Desorptionsenergieverläufen des Kupfers bildet sich allerdings bei etwa 0,85 ML ein Minimum aus, das zunächst im Rahmen der (BWA-) Näherung nicht erklärt werden kann. Seinen Ursprung hat dieses Minimum wahrscheinlich in einer weiteren 2D-Phasenumwandlung. Es handelt sich um den (Ordnungs-Ordnungs-) ps-cp-Übergang. Die Cu-Adteilchen verteilen sich nämlich bis zu einem bestimmten Bedeckungsgrad pseudomorph auf den vom Substrat vorgegebenen Adsorptionsplätzen an. Dieser bestimmte Bedeckungsgrad beträgt 0,87 ML (bezogen auf 1 ML bei eine dichte Packung der Adteilchen (cp), auf die der Begriff Monolage normiert ist). Bedingt durch den negativen misfit Cu-Re sind die Adteilchen dabei etwas expandiert auf dem Substratgitter angeordnet. Bei weiterem Ansteigen des Bedeckungsgrades verdichtet sich die Schicht, bis bei Q = 1 ML die dicht gepackte (cp-) Anordnung der Teilchen erreicht ist. Es ist nicht auszuschließen, daß dieser Effekt bereits lokal und zwar in großen Inseln auftritt, womit auch die Ausbildung des Minimums ab etwa 0,7 ML zu erklären wäre.
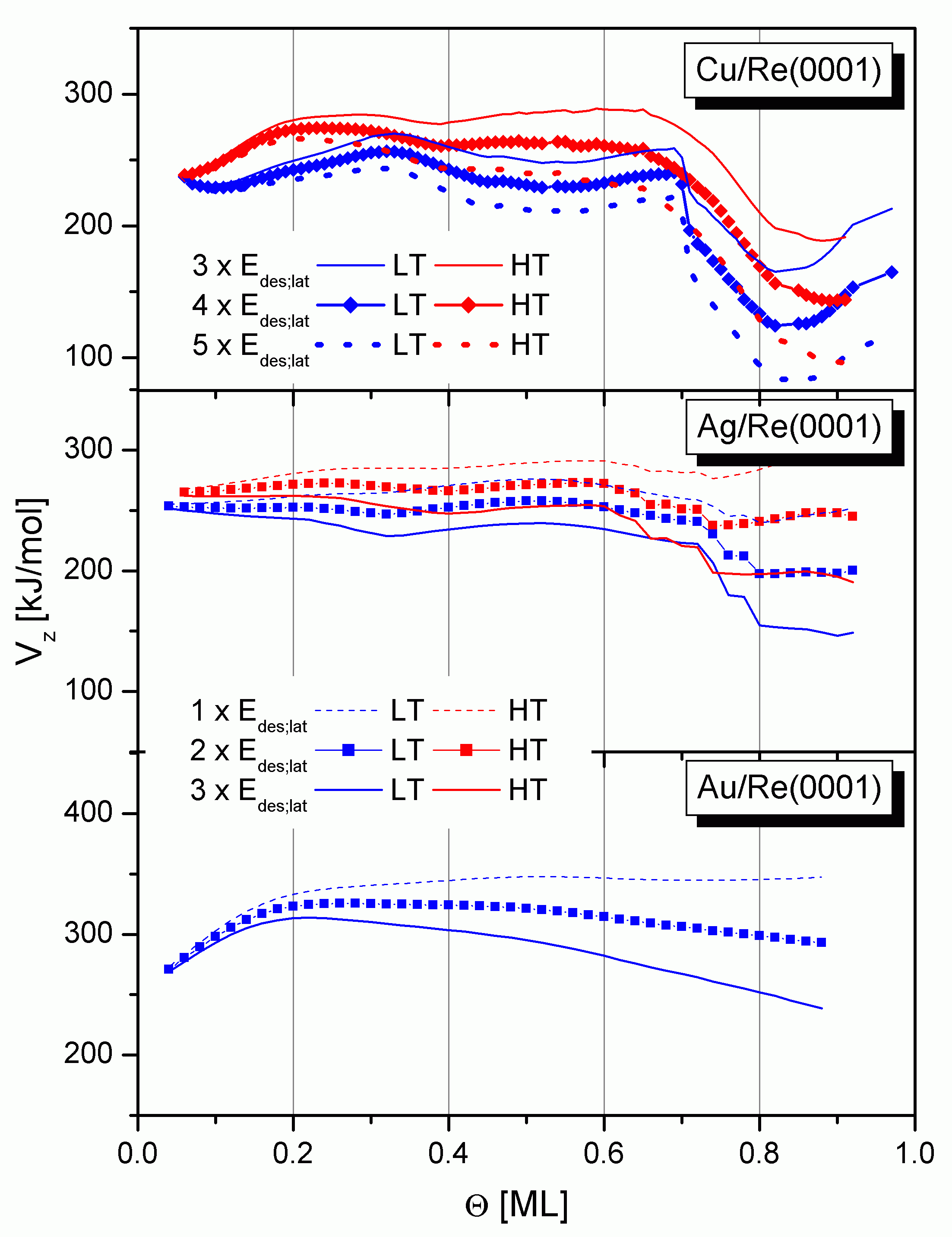
Das Absinken der experimentellen Desorptionsenergie als Folge des ps-cp-Übergangs deutet auf Veränderungen bzw. ein Absinken des z-Potentials Vz hin. Dieses Potential wurde bisher als additiver Anteil der Desorptionsenergie (
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
Für das Kupfer erkennt man den zunächst konstanten Verlauf der HT- und LT-Kurven bis 0,7 ML. Danach fällt Vz um ca. 80 kJ/mol ab. Dieser Abfall kann tatsächlich größenordnungsmäßig dem ps-cp-Übergang zugeordnet werden.
Beim Silber und beim Gold tritt dieser ps-cp-Phasenübergang im Submonolagenbereich noch nicht auf, sondern erst innerhalb der zweiten Lage. Auf Grund dessen kommt es auch in den Spektren nicht zur Ausbildung des Minimums. Bei diesen Systemen ist der misfit positiv, die Teilchen sind bei Q = 1 ML pseudomorph (und dabei komprimiert) auf dem Substrat angeordnet. Würde der ps-cp-Übergang bereits bei Q < 1 ML auftreten, würde sich der Abstand zwischen den Adteilchen vergrößern und der Bedeckungsgrad der ersten Lage verringern, d. h. Teilchen müßten in die zweite Lage übertreten.(Es könnte eventuell die um ca. 30 bis 50 kJ/mol niedrigere Desorptionsenergie beim Übergang von der ersten in die zweite Lage (vgl. auch
Beim z-Potential handelt es sich nicht nur um eine Wechselwirkung zwischen einzelnen Adsorbat- und Substratteilchen. Einen viel größeren Beitrag liefert bei Metall-auf-Metall-Systemen der Energiegewinn der Wechselwirkung der Wellenfunktion des Adsorbatteilchens mit der Gesamtwellenfunktion des Substrates (einschließlich der bereits vorhandenen Adsorbatinseln). Diese Wechselwirkung ist sehr stark davon abhängig, wie gut die Adteilchen optimale Substratplätze besetzen. Weichen sie von dieser Lage ab, so ergibt sich eine Schwächung der oben beschriebenen Wechselwirkung und damit eine Verringerung von Vz. Nach Rodriguez et al. kann es bei unterschiedlichen Elektronendichten von Adsorbat und Substrat nur dann zum optimalen charge transfer kommen, wenn auch optimale Gitterplätze besetzt sind [RoG92/1].
Gollish berechnete die folgenden Bindungspotentiale für die wichtigsten Adsorptionsplätze der Systeme Cu;Ag;Au/W(110) [Gol86/1]. Beim System Cu/W(110) ergeben sich daraus Energiedifferenzen zwischen den Adsorptionsplätzen von 19 bis 72 kJ/mol.
| Vz0 [kJ/mol] | Cu | Ag | Au |
| Mulde | 328 | 309 | 352 |
| on top | 256 | 245 | 270 |
| Brücke | 309 | 294 | 328 |
Wie bereits o. a. ist festzustellen, daß die berechnete lateralen Wechselwirkungsenergiebeträge nicht ausreichen, um die bedeckungsgradabhängigen Potentialverläufe Vz (Q) aus
| Cu | Ag | Au | |
| Vz0 [kJ/mol] | 270 | 260 | 310 |
| DVz(ps-cp) [kJ/mol] | -80 | -40 | -25 |
| EWW(real) [kJ/mol] | 19 | 8 | 10 |
Daß durch die Bragg-Williams-Näherung die laterale Wechselwirkungsenergie EWW zu klein wiedergegeben wird, ist bekannt [Hil62/b, Cla70/b]. Offenbar wirkt sich die komprimierte Anordnung der pseudomorphen Ag- bzw. Au-Schicht weniger stark auf die Wechselwirkungen der Adteilchen untereinander aus, als die expandierte Anordnung der ebenfalls pseudomorphen Cu-Schicht. Dies dokumentiert sich in den größeren Beträgen von DVz(ps-cp) und EWW(real) des Kupfers im Vergleich zum Ag und Au. Erklärt werden könnten die größeren Werte von EWW(real) gegenüber EWW mit der unsymmetrischen Form des Paar-Wechselwirkungspotentials (welches die bisher als konstant (mit dem Teilchenabstand) angenommene laterale Wechselwirkungsenergie ersetzt), das für Kompressionen einen steileren Verlauf hat als für Expansionen, vgl. auch qualitativ
|
|
|
Kapitel C4 | Abschnitt C - Münzmetallschichten auf Rhenium |
|
|
|
Kapitel C6 - Überblick: Cu, Ag und Au auf Re-Oberflächen |